Videos
Browse videos by topic
All Videos
Showing 1369-1392 of 1435 videos

IDMP: Why We Need It and How to Benefit
Epista Life Science
/@epistalifescience6136
Dec 6, 2016
The video provides a critical analysis of the necessity and benefits of implementing the ISO IDMP (Identification of Medicinal Products) standards within the life sciences industry. The speaker establishes that the drive for IDMP compliance is rooted in two primary motivations: a business ethic imperative and a legal requirement. Ethically, IDMP aims to address a significant public health crisis, citing data from the European Medicines Agency (EMA) indicating nearly 200,000 deaths annually in the European Union due to adverse drug events. By simplifying the identification of medicinal products and facilitating faster, clearer communication about drugs and their usage, IDMP is designed to accelerate adverse event tracking and ultimately lower this mortality rate. Legally, IDMP compliance is presented as non-negotiable for market access in the European Economic Area (EEA). The standard is a mandatory component of the new Pharmacovigilance Legislation, having been decreed by the European Commission in 2012. The speaker stresses that the question for pharmaceutical companies is not whether to adopt IDMP, but rather how to implement it, as continued market participation hinges on adherence. This regulatory push is framed as a "welcome pressure" because it forces organizations to confront and resolve deep-seated data management issues that often hinder operational efficiency and compliance. The core internal benefit of IDMP lies in its ability to drive organizational data cleanup and structuring. The speaker notes that in most companies, critical product information is fragmented, split across various departments, systems, and locations. This fragmentation leads to basic governance failures, such as the inability to definitively identify the owner, location, or most current version of simple attributes like a medicinal product’s name. IDMP requires the collection and standardization of over 90 specific product attributes. By demanding this level of detail and structure, the standard compels companies to consolidate their knowledge, moving away from unstructured, unknown, and unreliable data sources toward a centralized, structured, known, and reliable data environment. This transformation turns a regulatory burden into a strategic asset for business intelligence and operational excellence. Key Takeaways: * **Ethical Driver for Public Safety:** IDMP is fundamentally driven by the need to reduce the high incidence of adverse drug events, estimated to cause almost 200,000 deaths annually in the European Union, by enabling quicker and more reliable identification of medicinal products. * **Mandatory Legal Requirement:** Compliance with IDMP is a legal mandate under the new Pharmacovigilance Legislation, established by the European Commission in 2012, making it essential for any life science company wishing to maintain market presence in the EEA. * **Shift to Data-Centric Operations:** IDMP is a major catalyst forcing the life sciences industry to transition from outdated system-centric IT approaches—where data is siloed—to a modern data-centric approach that prioritizes data integrity and standardization across the enterprise. * **Data Fragmentation Challenge:** Companies struggle with basic data governance, often unable to pinpoint the exact location, current version, or responsible owner for simple product attributes (e.g., the medicinal product name), highlighting widespread internal data chaos. * **Welcome Pressure for Data Cleanup:** The regulatory requirement acts as a necessary force to compel organizations to collect, consolidate, and harmonize product information that is currently scattered across different departments and legacy systems. * **Scope of Standardization:** IDMP requires the rigorous standardization and structuring of more than 90 distinct attributes related to medicinal products, necessitating comprehensive data mapping and remediation projects. * **Strategic Business Benefit:** Beyond compliance, the IDMP implementation process offers a critical opportunity to clean up and summarize data, transforming unstructured, unreliable information into a structured, known, and reliable data source that can be leveraged for enhanced business intelligence. * **Improved Adverse Event Tracking:** Standardized identification and structured data enable regulatory bodies and companies to track any events related to pharmaceutical products much faster than was previously possible, enhancing pharmacovigilance capabilities. * **Data Ownership and Accountability:** The IDMP process forces organizations to clarify data ownership and accountability for specific attributes, resolving internal confusion regarding which department (e.g., marketing, regulatory affairs, manufacturing) is responsible for maintaining the accuracy of specific data points. Key Concepts: * **IDMP (Identification of Medicinal Products):** A set of ISO standards designed to standardize the identification and exchange of information on medicinal products globally, crucial for regulatory reporting and drug safety management. * **Pharmacovigilance Legislation:** The EU regulatory framework that mandates the implementation of IDMP to enhance the monitoring of drug safety and the reporting of adverse events post-market authorization. * **Data-Centric Approach:** A modern IT and operational strategy that focuses on data as the central asset, ensuring its quality, accessibility, and standardization across all systems and departments, in contrast to older system-centric models where data is often trapped within specific applications.

Intrinsic Clinical Systems - Using CRO Data to Automatically Feed Your CTMS
Intrinsic Clinical Systems
/@intrinsicclinicalsystems924
Nov 30, 2016
This video provides an in-depth demonstration of Intrinsic Clinical Systems, a Clinical Trial Management System (CTMS) developed by Pharmaca Consulting, focusing on its ability to efficiently integrate data, particularly from Contract Research Organizations (CROs), to automate CTMS updates and eliminate manual data entry. The presentation begins by outlining the common pain points associated with legacy CTMS solutions, which were often described as large, prohibitively expensive, packed with unused modules (like grants or drug supply), technologically antiquated, and requiring a heavy IT footprint due to varied platform architectures. The core mission of Intrinsic Clinical Systems was to address these issues by creating a light, modern, and easy-to-navigate system. The foundational architecture of the Intrinsic CTMS is built upon the Microsoft Dynamics operational platform. This strategic choice is highlighted as a key differentiator, providing native integration with the Microsoft Office Suite (Outlook, SharePoint, and Excel). This integration allows users to leverage familiar interfaces, utilize SharePoint for secure, HIPAA-compliant storage of eTMF documents, and view system reports directly within Outlook in real-time. The system is designed to be intuitive, requiring minimal training for users familiar with clinical trials, positioning it as a middle ground between expensive legacy systems and smaller, cloud-based startups. A significant portion of the demonstration focuses on data management, workflow, and integration capabilities. The system organizes clinical trials hierarchically (Project, Study, Study Country, Site) and employs role-based security (e.g., Global Admin, Study Manager, Read-Only Executive) to control user access and permissions, contrasting with user-based systems. To maintain high data standards, the system leverages Microsoft CRM’s duplicate detection settings, flagging similar entries for investigators or institutes based on criteria like email, phone number, and name characters, thereby preventing data duplication and misspelling across studies. Integration with external systems, particularly CROs, is achieved through three primary methods: building a direct API for sophisticated, real-time data transfer; utilizing a low-tech export/import feature via dynamic Excel templates for bulk updates; or setting up scheduled nightly data pulls from a designated SharePoint directory where CROs can upload files. Finally, the presentation covers the system’s Business Intelligence (BI) and reporting features, which utilize Microsoft Power BI. The system offers 10 out-of-the-box visualizations, including portfolio-wide views, study enrollment status reports, and site startup graphs. Users are also empowered with ad hoc reporting functionality by creating unlimited customizable "personal views" within the tabular data displays, allowing them to select and filter specific columns (e.g., budget information) and export these views as dynamic Excel worksheets that refresh automatically when opened. The system also confirms its regulatory focus, noting that its Trip Report functionality (covering routine monitoring, site initiation, and close-out visits) is actively undergoing 21 CFR Part 11 validation. Key Takeaways: * **Modern CTMS Architecture:** Intrinsic Clinical Systems is built on the Microsoft Dynamics operational platform, offering a light, modern alternative to legacy CTMS solutions that are often criticized for being expensive, feature-heavy, and technologically antiquated. * **Native Microsoft Integration:** The system provides seamless integration with the Microsoft Office Suite, enabling users to leverage familiar tools like Outlook (for real-time report viewing), Excel (for data import/export), and SharePoint (for HIPAA-compliant eTMF document storage). * **CRO Data Integration Methods:** The CTMS supports three distinct methods for integrating clinical data from CROs and other external systems: direct API builds for sophisticated real-time data flow, bulk data upload via dynamic Excel templates, and scheduled nightly data pulls from designated SharePoint directories. * **High Data Standards via Duplicate Detection:** The platform utilizes Microsoft CRM’s inherent duplicate detection settings to enforce data quality for critical entities like Investigators and Institutes, preventing study managers from entering duplicate or slightly misspelled records. * **Role-Based Security:** Access control is managed through predefined roles (e.g., Global Admin, Study Manager, Read-Only Executive), allowing organizations to precisely control what information different user groups can read, write, or access, rather than relying on complex user-by-user permissions. * **Ad Hoc Reporting Functionality:** Users can create unlimited custom "personal views" within the system’s tabular displays by easily adding or removing columns, effectively functioning as an ad hoc reporting tool to quickly visualize data points important to them (e.g., budget information). * **Dynamic Data Export:** Data can be exported as a dynamic Excel worksheet, which maintains a link to the CTMS database and automatically refreshes when opened, ensuring users always have the most current information without manual re-exporting. * **Power BI for Business Intelligence:** The system uses Microsoft Power BI for its out-of-the-box visualizations (10 available), providing comprehensive views of portfolio-level data, study enrollment status, and site startup metrics. * **Regulatory Compliance Focus:** The system is actively working toward 21 CFR Part 11 validation for its Trip Report functionality, which includes forms for routine monitoring, site qualification, site initiation, and close-out visits, ensuring compliance for electronic records and signatures. * **Simplified Workflow:** The system guides users through the clinical trial setup process using a clear hierarchy: Project, Study, Study Country, and Site, with mandatory fields kept minimal to allow for quick placeholder creation during the initial setup phase. Tools/Resources Mentioned: * Microsoft Dynamics (Operational Platform) * Microsoft Office Suite (Outlook, SharePoint, Excel) * Microsoft Power BI (Visualization Tool) * Spotfire (Mentioned as a comparable visualization tool) * Tableau (Mentioned as a comparable visualization tool) Key Concepts: * **CTMS (Clinical Trial Management System):** Enterprise software used to manage and track clinical trial operations, including site information, milestones, enrollment, and regulatory documents. * **eTMF (Electronic Trial Master File):** A digital repository for essential clinical trial documents, often integrated with or stored via the CTMS platform (Intrinsic uses SharePoint for this). * **21 CFR Part 11:** FDA regulation governing electronic records and electronic signatures, a critical compliance requirement for pharmaceutical software systems, particularly for documents like Trip Reports. * **General Entities:** System-wide data repositories (e.g., Investigators, Institutes, Projects) used for high-level viewing, navigation, and as a source of truth for specific studies. * **Specific Entities:** Information pertaining only to a particular record (e.g., Milestones or Trip Reports associated with a single Study or Site).

RocketWheel - Veeva - "Veeva Vault RIM Suite" - RocketWheel Explainer Videos
RocketWheel
/@Rocketwheel
Nov 30, 2016
This video provides an in-depth exploration of the challenges inherent in managing global regulatory information (RIM) and introduces the Veeva Vault RIM Suite as the solution for pharmaceutical and life sciences organizations. The presentation establishes the context by highlighting how supporting multiple markets makes regulatory information management highly complex. Everyday operational events, such as a manufacturing change or a health authority request, trigger extensive waves of necessary activities, including impact assessment, submission updates, and ensuring regional compliance across all affected territories. The analysis contrasts the inefficiencies of the "typical process" with the integrated capabilities of the Veeva Vault platform. Historically, regulatory processes are fragmented, relying on a patchwork of disparate systems. A typical workflow involves checking headquarters registration tracking, requesting information from affiliates, aggregating data in a separate project tracker, managing authoring and reviews in one system, publishing in another, dispatching submissions, locally tracking agency responses, and finally storing the massive dossier in a basic file share. This fragmentation results in critical information silos, making compliance extremely challenging and global visibility nearly impossible due to technological limitations. Veeva Vault RIM is presented as the technological evolution designed to eliminate these systemic issues by uniquely managing both content and data within a single, unified platform. This integration streamlines regulatory processes significantly. For example, a change event is now assessed within a global registration management system that directly links regulatory data to the original source documents. This ensures traceability and accuracy throughout the lifecycle. Furthermore, the system provides real-time tracking of submission documents and regulatory activities across all affected markets, offering one system for one comprehensive view across Regulatory departments globally. The ultimate goal is to enable organizations to move faster, gain critical visibility, and strengthen their overall compliance posture. Key Takeaways: • **Fragmentation is the Primary Barrier:** The traditional regulatory process is severely hampered by the use of separate, non-integrated systems for tracking, scheduling, authoring, publishing, and storage, which creates information silos and hinders efficient global operations. • **Trigger Events Demand Integrated Workflows:** Common operational events, such as manufacturing changes or health authority requests, necessitate complex, multi-regional regulatory workflows that require a unified system to manage the cascade of necessary assessment and submission activities. • **Compliance Risk from Poor Visibility:** Relying on project trackers and basic file shares makes it nearly impossible to maintain real-time visibility into the status of submissions and regional compliance, significantly increasing the risk of regulatory non-adherence. • **Veeva Vault RIM Unifies Content and Data:** The core value proposition of the Veeva Vault RIM Suite is its unique ability to manage both regulatory content (documents) and regulatory data (registrations, commitments) within a single platform, effectively eliminating the information silos that plague legacy systems. • **Global Registration Management:** The suite centralizes the assessment of change events using a global registration management system, which is crucial for ensuring that regulatory data is consistently linked back to the original source documents for auditability and traceability. • **Real-Time Tracking is Essential for Proactive Management:** The system provides real-time tracking of submission documents and regulatory activities across all affected markets, enabling regulatory teams to proactively manage deadlines and respond quickly to agency requests. • **Strengthening Regulatory Compliance:** By offering a single source of truth and automating the linkage between regulatory data and content, the solution directly contributes to strengthening compliance with critical regulations (e.g., FDA, EMA, GxP). • **Operational Efficiency Gains:** Streamlining the end-to-end process—from initial impact assessment and authoring through to publishing, dispatch, and tracking—allows life sciences companies to accelerate regulatory timelines and reduce manual administrative burdens. • **Single System for Global View:** Veeva Vault RIM establishes a "one system for one view" paradigm, ensuring that all stakeholders, regardless of geographic location, are operating from the same, current regulatory information, which is vital for global commercial success. Tools/Resources Mentioned: * Veeva Vault RIM Suite * Veeva Vault Key Concepts: * **Regulatory Information Management (RIM):** The comprehensive management of all data and documents related to regulatory submissions, product registrations, and compliance activities globally. * **Information Silos:** The isolation of data and content within separate, non-communicating systems, which prevents a holistic view of regulatory status and compliance. * **Global Registration Management:** The centralized process of tracking, updating, and managing product registrations and regulatory commitments across all international markets. * **Content and Data Integration:** The ability of a single platform (Veeva Vault) to manage both the structured data (e.g., registration dates, market status) and the unstructured content (e.g., submission dossiers, source documents) simultaneously.

RocketWheel - Veeva - "Veeva Vault Clinical Suite" - RocketWheel Explainer Videos
RocketWheel
/@Rocketwheel
Nov 13, 2016
The video introduces the Veeva Vault Clinical Suite, positioning it as the industry's first unified cloud platform designed to solve the chronic inefficiency and complexity associated with managing global clinical trials. The presentation begins by detailing the historical challenges faced by pharmaceutical and biotech companies, which have traditionally relied on a fragmented ecosystem of multiple, disconnected systems: one for creating study documents (like protocols), another for collecting site content (like CVs and financial documents), a third for driving study milestones (CTMS functions), and yet another for documenting regulatory compliance (eTMF functions). This reliance on siloed applications—often supplemented by spreadsheets, emails, and even physical mail—results in endless hours spent attempting to collate information to gain a full operational view of a single study or an entire program. The core problem identified is a technological limitation: application platforms historically managed either data (trial activities) or documents (content), but not both simultaneously. The Veeva Vault Clinical Suite overcomes this limitation by leveraging the Veeva Vault platform's unique ability to manage both data and content within a single system. This integration allows the suite to bring together Clinical Trial Management System (CTMS), electronic Trial Master File (eTMF), and Study Startup applications into "one process, one system, and one view across clinical operations." This unified approach is presented as the definitive method for eliminating information silos and streamlining complex clinical processes. The implementation of the unified suite provides immediate benefits across the clinical ecosystem. Sponsors, investigators, and Contract Research Organizations (CROs) all work within this single system, establishing a definitive single source of truth for trial content and data. Investigators benefit from real-time study updates, the ability to enter trial information once and leverage it across multiple sites and countries, and assurance that all required regulatory content is collected and current. For the sponsoring organizations, the unified view allows for rapid assessment of global trial status, immediate identification of site issues, and the ability to take corrective action quickly to meet study milestones, ultimately enabling faster, more informed decision-making and accelerating product time-to-market. Key Takeaways: • **Addressing System Fragmentation:** The primary operational bottleneck in clinical trials stems from the reliance on multiple, disconnected systems for managing content (e.g., protocols, regulatory documents) and data (e.g., milestones, site activities), leading to significant manual overhead and delays. • **The Unified Platform Value Proposition:** Veeva Vault Clinical Suite is highlighted as the first solution to integrate CTMS, eTMF, and Study Startup applications into a single system, providing a unified view across all clinical operations and eliminating the need for complex, error-prone data reconciliation between disparate platforms. • **Data and Content Cohesion:** The platform's unique ability to manage both operational data and regulatory content simultaneously is the foundational element that eliminates information silos, ensuring that documentation and trial activities are always synchronized. • **Enhanced Compliance and Audit Readiness:** By consolidating the eTMF (regulatory documentation) and CTMS (operational data) within one system, the platform inherently streamlines compliance tracking, automates audit trails, and ensures that the complete documentation required for GxP and regulatory submissions is readily accessible and current. • **Improved Stakeholder Collaboration:** The single-system approach mandates that sponsors, investigators, and CROs work within the same environment, fostering real-time collaboration, reducing communication latency, and ensuring all parties are operating from the same, validated source of truth. • **Real-Time Operational Intelligence:** Organizations gain the capability to quickly assess global trial status, proactively identify potential site issues or bottlenecks, and implement corrective actions faster, which is critical for meeting aggressive study milestones and accelerating trial execution. • **Increased Investigator Efficiency:** Investigators benefit from reduced administrative burden, receiving real-time study updates and only needing to enter trial information once, which the system then leverages across different sites and countries, minimizing redundant data entry and potential human error. • **Strategic Data Foundation for AI:** The establishment of a unified, clean data and content foundation within the Veeva Vault platform is essential for future AI and LLM initiatives, providing the structured, integrated data necessary for developing intelligent automation solutions for clinical operations and regulatory affairs. • **Focus on Global Complexity Management:** The solution is specifically designed to handle the complexity of global trials, which involves managing varying local requirements and functional area needs across different regions, emphasizing the necessity of a consistent, globally deployed platform. • **Accelerating Time-to-Market:** The ultimate goal of the unified suite is to accelerate the delivery of products to market by eliminating the operational friction and delays caused by fragmented information management, allowing companies to focus resources on scientific execution rather than administrative coordination. Tools/Resources Mentioned: * Veeva Vault Clinical Suite * CTMS (Clinical Trial Management System) * eTMF (electronic Trial Master File) * Study Startup Applications Key Concepts: * **Data and Content Unification:** The central concept of the Veeva Vault platform, referring to the ability to manage structured operational data (e.g., activity dates, site performance metrics) alongside unstructured content (e.g., protocols, consent forms, regulatory submissions) within a single technological infrastructure. * **Information Silos:** The traditional problem in clinical operations where critical information is stored in separate, non-communicating systems (e.g., spreadsheets, document repositories, activity trackers), leading to data fragmentation, redundancy, and difficulty in achieving a comprehensive view. * **Single Source of Truth:** A core data management principle achieved by the unified suite, ensuring that all stakeholders (sponsors, CROs, investigators) access the exact same, validated version of trial content and operational data, thereby improving decision-making accuracy.

Paul Attridge at 2016 Knowledge and Network Day
Epista Life Science
/@epistalifescience6136
Nov 11, 2016
This video provides an in-depth exploration of the compliance challenges and vendor assessment requirements facing the Life Science industry as it shifts toward a data-centric IT model utilizing regulated cloud environments. Paul Attridge, Senior Director at Veeva Vault R&D Europe, offers a vendor's perspective on how pharmaceutical and biotech companies can maintain data integrity and regulatory adherence while adopting cloud solutions. The core premise is that the cloud introduces new variables, but the fundamental need for rigorous supplier control remains paramount, stressing that cloud vendors must be treated exactly like any other general supplier providing solutions to a regulated environment. The presentation details several critical areas for customer due diligence. First, addressing the common anxiety around data residency, Attridge clarifies that customers must actively engage with vendors to ensure data is stored in a location suitable for their regulatory needs, countering the perception that cloud data is "just anywhere." Second, security is highlighted as a foundational requirement, specifically demanding dual-level encryption: data must be encrypted both while housed in storage (at rest) and while being transmitted across the internet (in transit). Customers must be able to test and verify the efficacy of both encryption layers to assure confidence in the security controls. The discussion also focuses heavily on vendor qualifications, emphasizing that a robust Quality Management System (QMS) is essential for any cloud provider in this space. The QMS must govern the vendor's environment, development processes, and infrastructure management to ensure consistency and compliance. Furthermore, given that cloud provision is a relatively new field, vendor experience is critical, as the provider must understand the business-critical nature of regulated data and the specific regulatory controls in place. Attridge concludes by emphasizing that the relationship with a cloud vendor is a long-term partnership built on trust, recommending that customers vet vendors based on verifiable certifications, accreditations, and their preparedness for emerging regulations such as the EU GDPR. Key Takeaways: * **Rigorous Vendor Assessment is Mandatory:** Customers must treat cloud vendors with the same level of control and scrutiny applied to traditional general suppliers, ensuring comprehensive oversight of their processes and infrastructure, especially when handling regulated data. * **Demand Specific Data Residency Confirmation:** Contrary to popular belief, cloud data location is not arbitrary. Customers must actively discuss and confirm with vendors that the physical location of their data storage meets all necessary regulatory and jurisdictional requirements. * **Verify Dual-Layer Encryption:** Security protocols must include encryption for data both at rest (while stored on servers) and in transit (while transmitted over the internet), and customers must have the ability to test and assure the efficacy of both layers. * **QMS is the Foundation of Compliance:** A vendor must possess a mature and verifiable Quality Management System (QMS) that manages their development lifecycle, infrastructure, and operational environment, which is vital for maintaining GxP compliance. * **Prioritize Experience in Regulated Business:** Because cloud technology is relatively new, customers must select vendors who demonstrate deep experience and understanding of the specific regulatory controls and business-critical nature of life sciences data. * **Leverage SOC Reports for Due Diligence:** Vendors must be prepared to provide Service Organization Control (SOC) reports, specifically Type 1 and Type 2, which enable customers to perform their own assessment of the vendor's internal controls and compliance posture. * **Assess Regulatory Foresight:** Vendors should demonstrate active preparation and adherence to evolving global regulatory frameworks, such as the upcoming EU GDPR regulations, ensuring their platform remains compliant with future standards. * **Trust is Built on Certifications:** Long-term trust is established through verifiable credentials. Customers should look for standard security certifications and accreditations that validate the vendor's quality management processes and security controls. * **Subscription Model Ensures Vendor Attention:** The subscription nature of cloud services means vendors must continuously work hard to retain the customer, often resulting in a high level of ongoing engagement and attention that customers should leverage to ensure service quality. Tools/Resources Mentioned: * Veeva Vault R&D (Example of a regulated cloud solution) * SOC Type 1 and Type 2 (Service Organization Control reports) Key Concepts: * **Data Integrity:** The core principle ensuring the accuracy, consistency, and reliability of data throughout its lifecycle, which is complicated by the transition to cloud infrastructure in regulated sectors. * **Regulated Cloud Environment:** The use of cloud services (SaaS, PaaS, IaaS) for handling GxP data, necessitating specific controls for security, validation, audit trails, and compliance with regulations like 21 CFR Part 11. * **Quality Management System (QMS):** The formal system used by vendors to manage their development, infrastructure, and operational processes to ensure consistent quality and regulatory adherence. * **Data-Centric IT Approach:** A strategic shift in IT infrastructure that prioritizes the management and sharing of data over the maintenance of individual, siloed systems, driven by modern regulatory requirements (e.g., IDMP, UDI). * **Encryption (At Rest and In Transit):** The essential security measure ensuring that sensitive data is protected both when stored on servers and when actively moving across networks.

How to Increase Efficiency of Clinical Trials with Content Management
USDM Life Sciences
/@usdatamanagement
Sep 13, 2016
This video provides an in-depth exploration of how to increase the efficiency of clinical trials through effective content management. Presented by Manu Vora, VP of USDM Life Sciences' Enterprise Content Management (ECM) practice, and Aaron Northington, VP of their Clinical practice, the discussion establishes that proper content management is critical for the success of clinical trials, especially given that many trials exceed budget and timelines. The presentation covers a range of topics from foundational best practices and strategic ECM implementation to innovative technological solutions and structured vendor selection. The speakers emphasize a pragmatic approach to content management, starting with simple, achievable goals and avoiding the pitfalls of over-customization. They highlight the transformative power of e-signatures and workflows in significantly reducing processing times and streamlining operations within and across business groups. Furthermore, the discussion delves into the importance of a holistic ECM strategy that considers not just technology, but also the people, processes, and content involved, ensuring that implementations are well-supported and aligned with organizational drivers. The video also touches upon leveraging industry standards, such as the DIA reference model for eTMF, to accelerate deployments and avoid common mistakes. It explores innovative solutions, including the use of structured content for regulatory integration, advanced feasibility survey systems, and the strategic adoption of cloud computing for enhanced collaboration and efficiency across the life sciences ecosystem. The session concludes with a detailed framework for ECM vendor selection, stressing the need to understand the entire content value chain—from creation to disposition—before committing to a solution. Key Takeaways: * **Criticality of Content Management in Clinical Trials:** Effective content management is paramount for clinical trial success. Statistics show that 7% of trials are over budget and exceed original timelines, and 20% of investigators recruit 80% of subjects. Well-structured, easily accessible content can significantly reduce timelines and accelerate trials. * **Adopt a "Crawl, Walk, Run" Approach:** When implementing clinical trial content solutions, start simple and avoid over-customization. Over-customizing systems leads to a high cost of ownership and makes future changes more challenging. Focus on establishing standard taxonomies and setting achievable goals, like having the first document in production by a set date. * **Leverage E-Signatures for Significant Time Savings:** E-signatures are a critical tool for optimizing clinical trial processes. Implementing e-signatures can drastically reduce the time required to obtain approvals on documents (e.g., from 19-58 days to 3-4 days for clinical trial documents), impacting areas like informed consent, investigator agreements, and visit reports. * **Implement Workflows for Process Streamlining:** Utilize workflows to automate and streamline processes within and across business groups. Examples include automating statistical programming approvals, quality reviews, or feasibility surveys for site startup, which can cut days, weeks, or even months off clinical trial timelines. * **Don't Reinvent the Wheel; Embrace Standards:** Avoid starting from scratch when setting up content management systems. Leverage industry-recognized models like the DIA reference model for eTMF taxonomy. This approach can prevent costly, failed implementations and accelerate progress, as demonstrated by a CRO that reduced eTMF deployment from 15-18 months to 3 months by adopting the DIA model. * **Holistic ECM Strategy is Essential:** A successful ECM implementation requires a comprehensive strategy that goes beyond just technology. It must consider the "people" (stakeholders, end-users, staffing, capabilities), "process" (policies, SOPs, work instructions, process remodeling workshops), and "content" (volume, governance, security, ownership) aspects to achieve efficient gains. * **Identify Key Drivers and Proactively Address Pain Points:** Understand the internal drivers for ECM (e.g., employee productivity, commercialization, scalability) and leverage solution enablers (e.g., single source of content access, process redesign). Be prepared for common pain points like complex legacy architecture, high IT investment costs, security risks, and process bottlenecks due to compliance. * **Utilize Robust Project Management Tools:** For complex ECM implementations, employ project governance models, baseline project timelines, and responsibility matrices (RACI) to ensure clear communication, alignment among numerous stakeholders (including external partners), and effective tracking of progress. * **Explore Innovative Clinical Content Solutions:** Consider solutions that integrate structured clinical trial content with regulatory information management systems to standardize global regulatory processes and accelerate submissions. Implement sound systems for feasibility surveys to create a maintainable database of potential sites. * **Embrace Cloud Computing for Cross-Organizational Efficiency:** Cloud capabilities offer a significant solution for streamlining business processes and resolving inefficiencies when working across multiple organizations (sponsors, CROs, vendors). This can enhance collaboration and accelerate clinical trials. * **Consider Specialized Cloud Solutions for Clinical Exchange:** Tools like Box, when GxP compliant, can serve as a clinical study exchange platform, facilitating cross-functional and external file transfers. This increases efficiency in communications, expedites site startup document exchange, and speeds up data transfer. * **Understand the Content Value Chain for Vendor Selection:** Before selecting an ECM vendor, thoroughly understand your content's entire value chain: creation (e.g., user ad-hoc, image capture, electronic forms), management (security, structure, review), operations (workflows, collaboration), retention (records management, archiving), and disposition. * **Implement a Structured Vendor Selection Approach:** Avoid rushing into an RFP. First, develop a clear ECM strategy by conducting needs and internal technology assessments, mobilizing teams, and defining the case for change. Only then proceed to solution design, RFP administration, vendor demos, and questionnaires. * **Available GxP Compliant Cloud ECM Systems:** Veeva is a leading compliant cloud solution in the life sciences space. Alfresco also offers cloud-based ECM. While platforms like Box and Dropbox provide cloud ECM, they are not currently GxP capable, but may be in the future. **Tools/Resources Mentioned:** * **DIA reference model for eTMF (Electronic Trial Master File):** A standardized model for managing clinical trial documents. * **Sprint methodology:** A five-day process for solving big problems and testing new ideas, as described in Jake Knapp's book "Sprint: How to Solve Big Problems and Test New Ideas in Just Five Days." * **Box:** Mentioned as a potential clinical study exchange platform for cross-functional and external file transfer (with a note on GxP compliance). * **Veeva:** Highlighted as a leading compliant cloud ECM solution in the life sciences industry. * **Alfresco:** Mentioned as a provider of cloud-based ECM solutions. * **SharePoint 365:** Mentioned as a tool for external collaboration. * **Project governance charts, project timelines, responsibility matrices (RACI):** Standard project management tools. **Key Concepts:** * **ECM (Enterprise Content Management):** A systematic approach to managing the lifecycle of information, from creation and storage to distribution and archiving, often within regulated environments. * **eTMF (Electronic Trial Master File):** An electronic system designed to manage all essential documents of a clinical trial, ensuring compliance, accessibility, and auditability. * **GxP:** A set of good practice regulations and guidelines (e.g., Good Clinical Practice, Good Manufacturing Practice) that ensure the quality, safety, and efficacy of products in regulated industries like life sciences. * **21 CFR Part 11:** Regulations from the U.S. Food and Drug Administration (FDA) that define the criteria under which electronic records and electronic signatures are considered trustworthy, reliable, and equivalent to paper records and handwritten signatures. * **Content Value Chain:** The complete lifecycle of content within an organization, encompassing its creation, management, use in business operations, retention, and eventual disposition. * **Dark Data:** Data that is acquired, processed, and stored but not used for any further purpose, leading to inefficiencies and missed opportunities. **Examples/Case Studies:** * A nationally renowned Clinical Research Institute successfully implemented an e-signature pilot program, reducing the time to obtain signatures on clinical trial documents from a range of 19 to 58 days down to just 3 to 4 days. * A large CRO (35,000 people) initially spent 15-18 months on an enterprise-wide eTMF implementation that failed due to over-customization and over-complication. After revamping their approach and adopting the DIA eTMF guidance, they successfully deployed the system in approximately three months.

Better Customer Data Means… Effective Compliance Activities
Veeva Systems Inc
@VeevaSystems
Jul 27, 2016
The video, produced by Veeva Systems, provides a focused analysis of the challenges life sciences companies face in managing aggregate spend data and ensuring compliance with global transparency guidelines. It establishes the context by highlighting the increasing complexity of operations in a global marketplace, which forces compliance officers, such as the fictional Sanjay at Vero bof farer, to reconcile expenditures for individual practitioners across multiple countries and disparate internal systems. The central problem is the difficulty in pulling together and harmonizing these multiple data sources, which introduces a high risk of delivering inaccurate information to health authorities, potentially resulting in regulatory penalties, fines, and significant damage to the company’s reputation. The core issue detailed in the video is the lack of a unified, accurate source for customer master data. When spend data—which includes payments, transfers of value, and other interactions that must be tracked for transparency reporting—is not tied to a uniquely identified professional profile, the reconciliation process becomes highly manual and prone to error. This fragmentation makes it nearly impossible for compliance teams to confidently track and report aggregate spend accurately, thereby failing to meet national and regional transparency requirements. The solution presented is the adoption of Veeva OpenData. The video positions OpenData as a foundational service that provides complete, accurate, and real-time customer data, including uniquely identified profiles for Healthcare Professionals (HCPs), organizations, and stewardship services across all major countries. By leveraging this centralized, verified data source, compliance officers can efficiently and accurately reconcile all spend data to a single, consistent customer identifier, regardless of the system or geography where the spend originated. Ultimately, the video argues that the switch to Veeva OpenData is essential for reducing the operational burden associated with managing compliant data. By ensuring data accuracy and unique identification, the solution allows life sciences companies to confidently comply with diverse national and regional transparency requirements, transforming a high-risk, fragmented process into a streamlined, reliable compliance activity. Key Takeaways: * **Global Compliance Challenge:** The complexity of tracking aggregate spend is amplified by the global nature of the life sciences marketplace, necessitating reconciliation of expenditures across diverse regulatory environments and multiple internal data systems. * **High Risk of Data Inaccuracy:** The primary risk in aggregate spend reporting stems from pulling data from non-harmonized sources, which often results in the submission of inaccurate information to health authorities, leading to potential regulatory action and reputational harm. * **The Necessity of Unique Customer Identification:** Effective and accurate transparency reporting relies fundamentally on the ability to uniquely identify and link spend data to specific Healthcare Professionals (HCPs) and organizations across all systems and geographic locations. * **Veeva OpenData as the Master Data Solution:** Veeva OpenData is presented as the critical tool for establishing a single source of truth, offering real-time, accurate profiles of customers and stewardship services necessary for reliable spend reconciliation. * **Harmonizing Transparency Reporting:** Utilizing a unified data foundation like OpenData enables life sciences companies to harmonize their transparency reporting processes, ensuring consistency and adherence to various national and regional guidelines simultaneously. * **Reducing Operational Burden:** Implementing a centralized data solution significantly reduces the manual effort required for data cleansing, verification, and reconciliation, thereby lowering the operational cost and time spent on compliance activities. * **Confidence in Regulatory Adherence:** The availability of complete and accurate customer data instills confidence in compliance officers that their reports meet regulatory standards, mitigating the risk of non-compliance penalties. * **Data Stewardship Services:** The mention of "stewardship services" indicates the importance of ongoing data maintenance and quality control, ensuring that customer profiles remain accurate and up-to-date in a dynamic global environment. Tools/Resources Mentioned: * Veeva OpenData Key Concepts: * **Aggregate Spend:** The total value of payments, transfers of value, and other expenditures made by pharmaceutical companies to or on behalf of healthcare professionals (HCPs) and healthcare organizations (HCOs). * **Transparency Guidelines/Regulations:** Regulatory mandates (e.g., the U.S. Sunshine Act, or similar global requirements) that require life sciences companies to publicly disclose aggregate spend data to ensure ethical interactions and prevent undue influence. * **HCP Profile Reconciliation:** The process of matching and consolidating spend data from various internal systems (e.g., ERP, CRM, expense systems) to a single, verified, and uniquely identified profile for a healthcare professional. Examples/Case Studies: * **Sanjay, Compliance Officer at Vero bof farer:** A fictionalized case study illustrating the common pain point of compliance officers struggling to reconcile global practitioner spend due to fragmented data sources, highlighting the need for a unified solution.

Better Customer Data Means…. More Engaging KOL Interactions
Veeva Systems Inc
@VeevaSystems
Jul 18, 2016
This video provides a focused case study demonstrating how accurate, centralized customer data is crucial for optimizing Key Opinion Leader (KOL) engagement within the pharmaceutical industry, specifically targeting the challenges faced by Medical Science Liaisons (MSLs). The narrative follows Andrew, an MSL at a fictional company, Vero BioPharma, who initially struggles with the time-consuming and inefficient process of aggregating KOL insights. The core problem identified is the fragmentation and lack of up-to-date information regarding KOLs, who often have complex profiles involving multiple roles and cross-border activities. This manual research prevents MSLs from focusing on high-value interactions, leading to wasted time and suboptimal meeting preparation. The presentation introduces Veeva OpenData and the Veeva KOL Data Subscription as the definitive solution to these operational inefficiencies. By switching to these integrated data sources, Andrew gains access to the most comprehensive and accurate healthcare provider (HCP) data across major countries. The KOL Data Subscription specifically enriches this foundational data with detailed insights necessary for strategic engagement. This centralization and enrichment capability directly addresses the complexity of tracking KOLs who frequently travel or hold diverse responsibilities across different geographies, enabling information reconciliation of all cross-border activities. The progression of the video highlights the transformation in the MSL workflow. Before the solution, Andrew spent "many hours every week" on basic research. After implementing the Veeva data solutions, he can now rely on real-time, accurate information at his fingertips, allowing him to manage an evolving stakeholder landscape effectively. The primary benefit is the ability to better tailor KOL interactions, shifting the MSL's focus from data aggregation to strategic engagement. The video concludes by positioning the switch to Veeva OpenData and KOL Data Subscription as essential for improving KOL identification and maximizing engagement quality, ensuring that valuable insights are not lost due and time is utilized efficiently. Key Takeaways: • **The MSL Data Challenge:** Medical Science Liaisons (MSLs) frequently lack access to comprehensive, centralized, and up-to-date KOL information, forcing them to spend significant time (potentially hours weekly) on manual research and aggregation of basic data points. • **Complexity of KOL Profiles:** Effective KOL engagement is hindered by the reality that these professionals often have complex, evolving profiles, including multiple roles, responsibilities, and cross-border activities, making manual tracking highly inefficient and prone to error. • **Veeva OpenData as the Foundation:** The solution relies on Veeva OpenData to provide the most comprehensive and accurate foundational healthcare provider (HCP) data across all major countries, serving as the single source of truth for commercial and medical teams. • **KOL Data Enrichment:** The Veeva KOL Data Subscription acts as an essential layer of enrichment, providing specialized and detailed KOL information necessary for strategic planning and tailoring interactions beyond basic contact details. • **Cross-Border Reconciliation:** A critical feature of the solution is its ability to reconcile information regarding cross-border activities, ensuring that MSLs have a complete view of a KOL's global influence and engagements, which is vital for global biopharma companies. • **Operational Efficiency Gain:** By centralizing and automating data aggregation, MSLs like Andrew are freed from time-consuming research, allowing them to reallocate their efforts toward higher-value activities, specifically focusing on deeper, more meaningful engagement with KOLs. • **Tailored Interactions:** Access to enriched, accurate data enables MSLs to tailor their interactions more effectively, ensuring the information shared is relevant to the KOL's specific roles, interests, and recent activities, thereby improving the quality and impact of the meeting. • **Stakeholder Landscape Management:** The integrated data solution allows pharmaceutical companies to proactively manage their evolving stakeholder landscape, ensuring that new or changing KOL roles and influence networks are immediately reflected in the system. • **Data Integrity for Compliance:** While not explicitly stated, the reliance on a single, validated source like Veeva OpenData inherently improves data integrity, which is crucial for maintaining regulatory compliance (e.g., managing consent, tracking interactions, and ensuring audit readiness). Tools/Resources Mentioned: * Veeva OpenData * Veeva KOL Data Subscription Key Concepts: * **Key Opinion Leader (KOL):** Highly influential healthcare professionals whose expertise and opinions significantly impact clinical practice and pharmaceutical strategy. * **Medical Science Liaison (MSL):** Field-based professionals within Medical Affairs responsible for building relationships with KOLs and communicating scientific information about products and therapeutic areas. * **Healthcare Provider (HCP) Data:** Comprehensive information regarding medical professionals, including contact details, affiliations, specializations, and prescribing habits.

Better Customer Data Means...More Efficient Commercial Operations
Veeva Systems Inc
@VeevaSystems
Jul 11, 2016
This video provides a focused, narrative-driven exploration of the critical challenges posed by poor customer data quality within pharmaceutical commercial operations and introduces Veeva OpenData as the solution. The narrative centers on Selma, the Sales and Marketing Director at Vero Biofarma, whose commercial effectiveness is severely hampered by inconsistent, outdated, and decentralized customer reference data. The presentation establishes that relying on disparate or poorly managed data directly undermines key functions such as sales force effectiveness, customer service, and direct marketing campaign execution, creating significant operational friction and financial waste. The core problem illustrated is the bottleneck created by legacy data management processes. Selma’s sales representatives submitted over 100 data change requests in a single month, yet these requests remained unaddressed by the data vendor, leading to frustration and inaccurate field execution. Furthermore, inconsistent data prevents the synchronization of help desk inbound calls with the CRM system, fragmenting the customer view. A specific financial consequence highlighted is a direct marketing campaign that ran significantly over budget due to the massive amount of duplicate and outdated customer records, demonstrating the tangible costs associated with poor data stewardship. Veeva OpenData is presented as the necessary paradigm shift, offering customer data that is rigorously verified, continuously updated, and centralized across all business functions and countries. The key differentiator emphasized is the superior structure and quality of the data, which ensures consistency globally. Crucially, the system drastically improves operational responsiveness by guaranteeing that data change requests submitted from the field are processed and turned around within a single business day. This rapid update cycle ensures that Selma and her teams have a consistent stream of timely, approved, and comprehensive customer data necessary for effective commercial execution, contrasting sharply with the weeks or longer required by traditional solutions. The video concludes by positioning Veeva OpenData as an essential tool for maintaining pace with constant market changes, ensuring that marketing, sales, and call center teams rely on the same source of truth for all customer interactions, campaigns, and support activities. The solution promises to eliminate the negative impacts of data inconsistency, duplication, and slow processing, thereby optimizing commercial operations responsiveness and maximizing the efficiency of field teams and marketing spend. Key Takeaways: • **Data Quality is a Commercial Bottleneck:** Poor customer data quality directly impedes commercial operations, leading to decreased Salesforce Effectiveness, fragmented customer service interactions (unsynchronized help desk calls with CRM), and inflated marketing campaign costs due to outdated or duplicate records. • **The Cost of Slow Data Stewardship:** The traditional model of data vendor management creates severe operational friction, exemplified by the scenario where over 100 field-submitted data change requests remained unaddressed for weeks, rendering field data inaccurate and frustrating sales teams. • **Operational Responsiveness is Key:** The speed of data change request processing is a critical metric for commercial success; legacy solutions requiring weeks for updates are unsustainable in a dynamic market, necessitating a shift to near real-time data governance. • **Veeva OpenData’s Value Proposition:** The primary benefit of Veeva OpenData is providing rigorously verified, continuously updated, and centralized customer data, ensuring a single source of truth across all global business functions. • **Rapid Data Turnaround:** Veeva OpenData significantly accelerates data stewardship, promising a turnaround time of just one business day for data change requests submitted by the field, which directly supports timely and accurate commercial execution. • **Impact on Marketing ROI:** Outdated and duplicate customer data leads to massive waste in direct marketing campaigns; implementing a solution like OpenData is essential for cleaning lists, reducing unnecessary spend, and improving campaign targeting and ROI. • **Need for Data Consistency:** Commercial teams (marketing, sales, call centers) must rely on the exact same customer data for campaigns, interactions, and support to ensure a unified customer experience and accurate reporting on commercial activities. • **Data Structure and Quality:** The solution emphasizes consistently superior data structure and quality, which is crucial for seamless integration with CRM systems and downstream business intelligence tools used for actionable insights. • **Centralized Data Governance:** Centralizing customer data across all business functions and countries is vital for large pharmaceutical organizations to ensure global compliance and consistent commercial messaging. Tools/Resources Mentioned: * Veeva OpenData * Veeva CRM (implied as the system receiving the data) Key Concepts: * **Salesforce Effectiveness (SFE):** A measure of how efficiently and effectively a sales organization achieves its objectives, which is directly dependent on the quality and timeliness of customer data available to reps. * **Customer Reference Data:** The foundational, standardized data set (names, addresses, affiliations, specialties) used to identify and interact with healthcare professionals (HCPs) and organizations (HCOs). * **Data Change Request (DCR):** A formal request submitted by a field user (e.g., a sales rep) to update or correct inaccurate information within the customer reference data system. * **Commercial Operations:** The function within a pharmaceutical company responsible for supporting sales, marketing, and customer service activities, often relying heavily on CRM and data management systems.

Better Customer Data Means…More Productive Reps
Veeva Systems Inc
@VeevaSystems
Jul 6, 2016
This video provides an illustrative case study demonstrating the critical importance of accurate customer reference data for pharmaceutical commercial operations, specifically focusing on the challenges faced by field sales representatives and the strategic solution offered by Veeva OpenData. The narrative centers on Selma, a Sales and Marketing Director at Virtio Biopharma, whose team repeatedly struggles with operational inefficiencies and missed opportunities stemming from poor data quality. The core problem is vividly illustrated through a common field scenario: one of Selma’s sales representatives visited St. Vincent’s Hospital intending to see Dr. Bowen, only to discover the physician had recently moved to a new practice in another city. This situation resulted in immediate negative consequences: the rep was embarrassed, appeared unprofessional to hospital staff, and wasted valuable time and resources. Furthermore, the outdated system prevented the rep from identifying Dr. Bowen’s replacement or appropriately documenting the outcome of the failed visit within their system. This recurring issue led Selma to lose faith in the trustworthiness of the foundational data driving her team's customer interactions, directly impacting commercial effectiveness. The video positions Veeva OpenData as the necessary solution to restore data integrity and optimize field force performance. By switching to this platform, Selma and her sales representatives gain real-time access to a comprehensive, full database of customer information. This new approach immediately addresses the pain points of inaccurate targeting and wasted effort. The improved data quality ensures that reps are better equipped with the correct customer details, allowing them to eliminate unproductive calls and visits to incorrect targets. The ultimate outcome is a significant boost in rep productivity, reinforced trust in the underlying data infrastructure, and the ability for the commercial team to focus exclusively on meaningful engagement with the right healthcare professionals (HCPs). The presentation concludes by highlighting Veeva OpenData's attributes as being "open, easy, and global," positioning it as the standard for maintaining high-quality customer reference data in the life sciences sector. Key Takeaways: • **Data Inaccuracy Cripples Commercial Efficiency:** The failure to maintain up-to-date customer reference data (such as HCP location or status changes) directly results in wasted resources, including rep time, travel costs, and missed opportunities for meaningful engagement. • **Rep Productivity is Directly Tied to Data Quality:** When field representatives encounter outdated information, their productivity plummets because they spend valuable selling time correcting data errors or traveling to incorrect locations, rather than interacting with target customers. • **The Rep Experience Impacts Professionalism:** Arriving at a location only to find the target HCP has moved makes the sales representative appear unprofessional and unprepared, potentially damaging the company's reputation with office staff and future interactions. • **Data Trust is Essential for System Adoption:** When sales directors like Selma lose faith in the trustworthiness of the data driving their CRM and commercial systems, it undermines the entire technology investment and leads to reduced user adoption and reliance on manual workarounds. • **Documentation Requires Accurate Reference Data:** The inability to accurately document visit experiences, find replacement contacts, or update system records due to poor reference data compromises regulatory compliance tracking and future strategic planning. • **Veeva OpenData Solves the Reference Data Challenge:** The platform is presented as a solution that provides real-time access to a comprehensive customer database, ensuring that field teams are always equipped with the most current information. • **Focus Shift from Logistics to Engagement:** By eliminating the logistical hurdles caused by bad data, accurate reference data allows sales representatives to shift their focus entirely onto high-value activities: interacting with the right customers and delivering relevant information. • **The Need for Global, Accessible Data:** The solution emphasizes that high-quality customer data must be "open, easy, and global," suggesting that pharmaceutical companies require a standardized, easily integrated data source that works across different territories and commercial systems. Tools/Resources Mentioned: * **Veeva OpenData:** A customer reference data solution designed specifically for the life sciences industry, providing accurate, real-time information on healthcare professionals (HCPs) and organizations. Key Concepts: * **Customer Reference Data:** The foundational, non-transactional data about healthcare professionals (HCPs), organizations, and their affiliations, which is crucial for accurate targeting, segmentation, and compliance within the pharmaceutical commercial model. * **Rep Productivity:** A measure of the efficiency and effectiveness of field sales representatives, often quantified by the number of successful, documented interactions with target HCPs versus time spent on administrative tasks or failed calls. * **Commercial Operations Data Trust:** The confidence that commercial leadership and field teams have in the accuracy and reliability of the data residing within their CRM and business intelligence systems, which is necessary for strategic decision-making and efficient execution.

TMF/eTMF Regulatory Agency Expectations, Inspections, and Findings Trailer
Kathy Barnett
/@kathybarnett4070
Jun 21, 2016
This video provides an in-depth exploration of Trial Master File (TMF) and electronic Trial Master File (eTMF) regulatory expectations, common inspection findings, and strategies for effective corrective and preventive actions (CAPAs). The speaker, Donna Dorzinski, leverages 26 years of experience in big Pharma and regulatory compliance consulting, emphasizing her active role in the TMF reference model working group. The presentation aims to equip attendees with a clear understanding of current regulatory demands from agencies like MHRA, EMA, and FDA, enable them to identify prevalent TMF/eTMF-related findings, and provide actionable strategies for proactive compliance and successful resolution of inspection issues. The core of the discussion revolves around the evolving definition and scope of the TMF. Historically, the TMF was often narrowly perceived as a collection of "essential documents" primarily focused on clinical aspects. However, the speaker emphasizes that the TMF, as defined by the European directive from 2005, is a standalone set of documentation that should not require additional explanation from staff. It must comprehensively allow for the evaluation of trial conduct, data integrity, and compliance with Good Clinical Practice (GCP). This means the TMF must "tell the story" of the study, reflecting everything that happened, rather than just being a checklist of documents. A critical point is that the TMF is a collective output from *all* functional areas involved in a clinical trial, extending beyond clinical to include data management, biostatistics, clinical trial material management, and pharmacovigilance. The video further delves into the implications of the ICH E6 integrated addendum, released prior to the seminar, which introduced a crucial requirement: sponsors and investigators must maintain a record of the *locations* of their respective essential documents. This addresses the reality that not all TMF content resides within a single "TMF" or "eTMF" system, citing pharmacovigilance databases as an example for safety documentation. The speaker clarifies that knowing the location of a record is sufficient to meet regulatory requirements, provided the storage system allows for easy identification, search, and retrieval, regardless of media (paper, digital, cloud). Moreover, ICH E6 acknowledges that individual trials may necessitate additional documents beyond the traditional "essential document list," reinforcing the broader, more comprehensive view of the TMF as a complete narrative of the study. The ultimate objective is to enable organizations to prepare in advance, putting processes in place to prevent regulatory findings related to TMF management. **Key Takeaways:** * **Evolving Regulatory Scrutiny on TMF/eTMF:** Regulatory bodies like MHRA and EMA have significantly heightened their focus on TMFs. MHRA explicitly defines TMF deficiencies (e.g., unavailability, inaccessibility, incompleteness) as critical Good Clinical Practice (GCP) inspection findings, underscoring the severe consequences of non-compliance. * **TMF as a Standalone Narrative:** The TMF must function as a comprehensive, standalone set of documentation that fully narrates the conduct of a clinical trial. It should enable the evaluation of trial conduct, data integrity, and GCP compliance without requiring supplementary verbal explanations from staff, which is vital during inspections, especially if key personnel are unavailable. * **Beyond "Essential Documents":** The traditional, narrow interpretation of the TMF as solely a collection of ICH E6 Section 8.1 "essential documents" is outdated and insufficient. The TMF must encompass all records that genuinely reflect the entire study process, extending beyond clinical documentation to include contributions from data management, biostatistics, clinical trial material management, and pharmacovigilance. * **ICH E6 Integrated Addendum's Impact:** The recent ICH E6 integrated addendum (released prior to the seminar) introduced a critical mandate: sponsors and investigators must meticulously maintain a record of the *locations* of their essential documents. This update acknowledges the distributed nature of TMF content across various systems and databases, such as pharmacovigilance databases for safety data. * **Location Knowledge is Key for Compliance:** Regulatory compliance for TMF content does not necessarily demand the duplication of documents across multiple systems. As long as an organization can precisely identify the location of a record and ensure its easy search and retrieval, this satisfies the regulatory requirement for documentation management. * **Accessibility and Retrievability are Paramount:** Irrespective of the storage medium (paper, digital, or cloud), the TMF system must guarantee effortless identification, searching, and retrieval of documents. The ability to quickly locate and present requested documentation is a non-negotiable factor for navigating regulatory inspections successfully. * **Proactive CAPA Strategies:** The seminar strongly advocates for developing effective Corrective and Preventive Actions (CAPAs) that not only address existing regulatory findings but also proactively implement robust processes to prevent future occurrences. This forward-thinking approach is crucial for achieving consistently successful inspection outcomes. * **Cross-Functional TMF Ownership:** The TMF is inherently a collective output from all functional areas involved in a clinical trial. This necessitates a collaborative, cross-functional approach to TMF management, ensuring that every relevant department contributes to and maintains its documentation in a compliant, accurate, and accessible manner. * **Anticipate Additional Documentation Needs:** ICH E6 explicitly acknowledges that individual trials may necessitate documents beyond the standard "essential document list." Organizations must be prepared to include any additional documentation required to comprehensively reflect the conduct and integrity of a specific trial. * **Importance of Robust Site Records:** The speaker's anecdote about a successful site inspection despite the coordinator's absence underscores the critical importance of meticulously maintained, standalone site records, regulatory documentation, and ethics documentation. Such diligence ensures operational continuity and compliance even in unforeseen circumstances. **Key Concepts:** * **Trial Master File (TMF):** A comprehensive collection of documents that individually and collectively permit the evaluation of the conduct of a clinical trial, the quality of the data produced, and compliance with GCP. It must be a standalone set of documentation that tells the complete story of the study. * **Electronic Trial Master File (eTMF):** The digital version of the TMF, storing all trial-related documents electronically. * **Good Clinical Practice (GCP):** An international ethical and scientific quality standard for designing, conducting, recording, and reporting trials that involve the participation of human subjects. * **Corrective and Preventive Actions (CAPA):** A systematic process for identifying and addressing existing nonconformities (corrective actions) and preventing their recurrence (preventive actions), particularly in response to TMF-related regulatory findings. * **ICH E6 (R2) Integrated Addendum:** An update to the International Conference on Harmonisation (ICH) guideline for Good Clinical Practice, providing additional guidance on quality management, risk-based monitoring, and electronic systems, including specific requirements for documentation location. * **Essential Documents:** As defined in ICH E6 Section 8.1, these are documents that permit the evaluation of the conduct of the trial and the quality of the data produced, demonstrating compliance with GCP and regulatory requirements. The video clarifies that the TMF extends beyond this list. **Examples/Case Studies:** * **Successful Site Inspection with Absent Coordinator:** The speaker shared a real-world example of a site inspection that proceeded smoothly and successfully despite the study coordinator being on medical leave and unreachable. This success was directly attributed to the coordinator's meticulous maintenance of standalone site records, regulatory documentation, and ethics documentation, highlighting the critical value of a well-prepared TMF for business continuity. * **"Deer in the Headlights" Scenario:** A common pitfall during regulatory inspections is when an inspector requests a specific document, and the staff is unable to locate it or the available documentation fails to adequately answer the regulator's question. This scenario underscores the absolute necessity for a TMF that is easily searchable, retrievable, and comprehensive enough to stand alone and address all potential inspector queries. * **Pharmacovigilance Database as TMF Content Location:** The speaker used pharmacovigilance databases, where safety documentation is frequently stored, as a prime example of TMF content that may reside outside the primary "TMF" or "eTMF" system. This illustrates the practical application of the ICH E6 addendum's requirement to know the *location* of documents rather than mandating their duplication across systems.

eTMF Implementation Trailer
Kathy Barnett
/@kathybarnett4070
May 17, 2016
This video provides an in-depth exploration of implementation strategies for an electronic Trial Master File (eTMF) within the pharmaceutical and life sciences industry. The speaker, Donna Dorzinski, an industry veteran with 26 years of experience, including 15 years in big pharma clinical operations and 11 years as president of Justin time gcp, frames the transition to eTMF as a significant opportunity for business process improvement. She emphasizes that while implementing an eTMF can be challenging, it forces organizations to critically examine their existing processes and optimize them, ultimately leading to a higher quality TMF that ensures inspection and audit readiness. The presentation delves into critical considerations for a successful eTMF rollout, moving beyond just the technical aspects to encompass organizational and strategic elements. A core theme is the necessity of broad stakeholder engagement, highlighting that eTMF implementation cannot occur in isolation. The speaker outlines strategies for effective communication with business partners and for addressing the impact on various functional areas beyond clinical operations, as the eTMF touches many parts of an organization. A practical approach is suggested for developing user requirements, which are crucial for informed vendor selection. Further, the video details key technical and procedural aspects that underpin a robust eTMF system. It stresses the importance of adopting a standard indexing structure, advocating for the DIA (Drug Information Association) reference model as it rapidly becomes an industry standard. A significant portion of the discussion is dedicated to the strategic use of metadata, explaining its power in searchability, record identification, quality control, and operational enhancement. However, a crucial caveat is provided: organizations must be judicious in selecting metadata to track, ensuring that each piece adds value and is genuinely used for searching or business insights, rather than creating unnecessary work. The presentation concludes by emphasizing the establishment of clear conventions for file naming and record filing to maximize the organization and value of the eTMF. Key Takeaways: * **eTMF as a Catalyst for Process Improvement:** Implementing an eTMF should be viewed not merely as a technology upgrade but as a strategic opportunity to review and improve existing business processes across the organization, leading to greater efficiency and quality. * **Mandatory Stakeholder Engagement:** Successful eTMF implementation requires extensive communication and collaboration with all business partners and functional areas, as the system's impact extends far beyond clinical operations. It cannot be implemented in a vacuum. * **User Requirements Drive Vendor Selection:** A critical first step is to develop a comprehensive list of user requirements. These requirements will serve as the foundation for evaluating and selecting an eTMF vendor that best aligns with the organization's specific needs and operational workflows. * **Standardized Indexing is Key:** Adopting a standard indexing structure, such as the DIA reference model, is crucial for consistency, searchability, and industry alignment. The DIA reference model is rapidly becoming the industry standard and facilitates better organization and interoperability. * **Strategic Use of Metadata:** Metadata is a powerful tool for enhancing eTMF searchability, identifying specific records, performing quality control, and gaining insights. Organizations should thoughtfully select metadata types that directly support their business operations and search needs. * **Avoid Redundant Metadata Collection:** While metadata is powerful, it's vital to collect only metadata that adds genuine value and will be actively used for searching or operational enhancement. Collecting excessive, non-valuable metadata can create significant extra work without providing commensurate benefits. * **Establish Clear Conventions:** To maximize the value and usability of an eTMF, organizations must establish clear conventions for naming files and filing records. Well-defined conventions ensure consistency, improve organization, and make the TMF more accessible and auditable. * **Regulatory Readiness as a Primary Driver:** A high-quality eTMF system is essential for ensuring an organization is inspection and audit ready, meeting regulatory requirements from bodies like the FDA and EMA. This focus on compliance is a significant benefit and driver for eTMF adoption. * **Impact Across Functional Areas:** eTMF implementation affects various functional areas within an organization, not just clinical operations. It's important to consider and address these broader impacts during the planning and execution phases. * **Collaboration with CROs and Sponsors:** The implementation process must account for interactions and collaboration with Contract Research Organizations (CROs) and sponsor partners, ensuring seamless integration and data exchange within the eTMF ecosystem. Tools/Resources Mentioned: * **DIA Reference Model:** The Drug Information Association (DIA) reference model for the Trial Master File, highlighted as a rapidly emerging industry standard for TMF indexing. Key Concepts: * **eTMF (electronic Trial Master File):** A digital system for managing and storing essential documents and records related to a clinical trial, replacing traditional paper-based TMFs. * **TMF (Trial Master File):** A collection of essential documents that individually and collectively permit the evaluation of the conduct of a clinical trial and the quality of the data produced. * **Metadata:** Data that provides information about other data. In an eTMF, metadata can include details like document type, study phase, date, author, or specific therapeutic area, enabling powerful search and organization capabilities. * **Indexing:** The process of organizing and categorizing documents within the TMF, often following a standardized structure to ensure consistency and ease of retrieval. * **Inspection Readiness:** The state of being prepared to demonstrate compliance with regulatory requirements during an inspection by regulatory authorities (e.g., FDA, EMA). * **Audit Readiness:** The state of being prepared to demonstrate compliance with internal policies, procedures, and external regulations during an audit.

Trial Master File for Sponsors Trailer
Kathy Barnett
/@kathybarnett4070
Apr 20, 2016
This video provides an in-depth exploration of the Trial Master File (TMF) for sponsors, focusing on its setup, maintenance, and critical role in clinical trial oversight. The speaker, a consultant with over 25 years of experience in the pharmaceutical industry and a contributor to the TMF Reference Model, frames the discussion around the evolving regulatory landscape. She emphasizes that TMF management has significantly changed in the last five to ten years, making it a relevant topic for both seasoned professionals and newcomers to the industry. The session aims to offer a "30,000-foot flyover" of essential TMF concepts, policies, and quality control measures. The presentation delves into the rationale behind the TMF, defining it as the comprehensive "diary" or "story" of a clinical trial from its inception to conclusion. The speaker highlights that the TMF is explicitly referenced and required by major regulatory bodies, including the Code of Federal Regulations, EU Directives, and ICH guidelines, underscoring its universal importance in demonstrating compliance. A key concept discussed is that the TMF must be a "standalone set of documentation" capable of telling the entire story of the trial without requiring additional explanation from the study team. This ensures that regulatory inspectors can independently evaluate the conduct of the clinical trial, the integrity of the data, and adherence to Good Clinical Practice (GCP). Furthermore, the video addresses the practical aspects of TMF management, including its required components, policy recommendations, and the crucial activities of maintenance, quality control, and quality assurance. The speaker advocates for companies to develop their own tailored TMF policies and procedural documents rather than relying on generic templates, stressing that each organization's practices are unique. She notes the shift in industry perception, where TMF is now recognized as a multi-disciplinary responsibility extending beyond just clinical departments. The discussion also touches upon the transition from traditional paper-based TMFs to electronic TMF (eTMF) systems, acknowledging that while many companies are adopting eTMF, paper systems are still prevalent. The core principle reiterated throughout is: "if it isn't documented, it didn't happen," or more precisely, "if you don't have access to the documentation, it didn't happen," emphasizing the critical need for accessible and complete records. Key Takeaways: * **Evolving Regulatory Landscape:** The management of Trial Master Files (TMFs) is not static; the regulatory climate has undergone significant changes in the last five to ten years, necessitating continuous adaptation in how TMFs are managed and maintained. * **TMF as the Trial's Narrative:** The TMF serves as the complete "diary" or "story" of a clinical trial from beginning to end, providing a comprehensive record of all activities and decisions, which is essential for demonstrating accountability and transparency. * **Universal Regulatory Mandate:** TMFs are explicitly required and referenced across major regulatory bodies, including the Code of Federal Regulations (CFR), EU Directives, and ICH guidelines, highlighting their fundamental importance in global clinical research. * **Standalone Documentation Principle:** A TMF must function as a standalone set of documentation, meaning it should be self-explanatory and not require additional verbal explanation from the associated sponsor or staff, enabling independent evaluation by inspectors. * **Evaluation of Compliance and Data Integrity:** The primary purpose of the TMF is to allow regulatory inspectors to evaluate whether a study was conducted in compliance with Good Clinical Practice (GCP) and if the data possesses the integrity required for compound or device approval. * **Multi-Disciplinary Responsibility:** TMF management is no longer solely the responsibility of clinical departments; it is a multi-disciplinary effort that involves outputs and contributions from various functional areas within the sponsor organization. * **Essential Documents Defined:** The term "essential documents" is synonymous with the TMF, encompassing all documentation necessary to permit the evaluation of the trial, assess data quality, and confirm compliance with GCP and regulatory requirements. * **Tailored Policy Development:** Companies should establish their own robust TMF policies and procedural documents, customized to their specific practices and processes, rather than relying on generic Standard Operating Procedures (SOPs). * **Focus on Maintenance and Quality Control:** Effective TMF management requires diligent maintenance, comprehensive quality control (QC), and quality assurance (QA) activities to ensure the accuracy, completeness, and accessibility of documents. * **Shift Towards Electronic TMF (eTMF):** There is a clear industry trend towards the adoption of electronic TMF systems, moving away from traditional paper-based methods, though a significant number of companies still utilize paper TMFs. * **TMF Scope Beyond ICH E6:** Modern TMFs are more extensive than the requirements outlined in ICH E6 alone, making their maintenance increasingly complex and necessitating comprehensive strategies that go beyond basic compliance. * **Fundamental Principle of Documentation:** The core tenet "if it isn't documented, it didn't happen" (or "if you don't have access to the documentation, it didn't happen") underscores the critical importance of meticulous and accessible record-keeping in clinical trials. * **TMF Reference Model Contribution:** The speaker is actively involved with the TMF Reference Model group, having led revisions to Zone 4 (Ethics Committee review), indicating the model's significance as an industry standard for TMF structure. Key Concepts: * **Trial Master File (TMF):** A collection of essential documents that individually and collectively permit the evaluation of the conduct of a clinical trial, the quality of the data produced, and compliance with Good Clinical Practice (GCP) and regulatory requirements. * **Electronic Trial Master File (eTMF):** A digital system used for the management, storage, and archiving of TMF documents, offering advantages in accessibility, searchability, and compliance. * **Good Clinical Practice (GCP):** An international ethical and scientific quality standard for designing, conducting, recording, and reporting trials that involve the participation of human subjects. * **Essential Documents:** All documents that individually and collectively permit the evaluation of the conduct of a clinical trial and the quality of the data produced. These are effectively the contents of the TMF. * **TMF Reference Model:** An industry-standard, hierarchical model for structuring and organizing TMF documents, designed to improve consistency and facilitate compliance. Tools/Resources Mentioned: * **TMF Reference Model:** An industry-developed standard for organizing and managing Trial Master File documents.

eTMF Quality Oversight: A Risk-Based Approach Trailer
Kathy Barnett
/@kathybarnett4070
Mar 31, 2016
This video provides an in-depth exploration of eTMF (electronic Trial Master File) quality oversight, emphasizing a risk-based approach to ensure inspection readiness and overall Good Clinical Practice (GCP) compliance. The speaker, drawing from over 25 years of experience in large pharmaceutical companies and as an industry consultant, highlights the critical role of a high-quality TMF as the sole evidence during regulatory inspections. The session aims to equip attendees with insights into building a risk-based assessment plan for TMF quality control (QC) activities and identifying high-risk artifacts that commonly lead to quality issues. The presentation establishes that a regulatory inspection's success is directly tied to the quality and completeness of the TMF. While verbal explanations can clarify, only documented evidence within the TMF can substantiate claims made to regulators. This foundational principle underscores the necessity of proactive TMF management, where inspection readiness is integrated from the study's inception rather than being a last-minute scramble. The speaker shares personal experiences of the "scrambling feeling" when trying to locate documents during an inspection, reinforcing the value of a well-maintained TMF. Key topics covered include the application of risk-based assessment to structure TMF QC activities, various methods to ensure a high-quality TMF, and a detailed discussion of specific documentation "artifacts" that frequently pose quality risks. The speaker defines the TMF, referencing the European Directive 2005, as a standalone set of documentation that should tell the complete story of a study without requiring extensive additional explanation from sponsor or site staff. This self-sufficiency is crucial given the inevitable team changes throughout a study's lifecycle. Furthermore, the video stresses that while clinical groups often "own" the TMF, all functional areas contributing content bear responsibility for its quality, extending beyond clinical operations to data management and statistics. The speaker also notes experience with the TMF Reference Model, having chaired a revision, and significant experience with 21 CFR Part 11 concerning the validation of clinical and TMF systems. The overarching message is that a quality TMF is one that is complete, timely, and comprised of high-quality records, demonstrating that a study was conducted in accordance with GCP requirements and ensuring data integrity. By implementing a risk-based approach to TMF oversight and QC, organizations can systematically identify and mitigate potential quality issues, thereby ensuring their TMF is robust, reliable, and fully prepared for regulatory scrutiny. Key Takeaways: * **TMF as Primary Evidence:** The Trial Master File (TMF) serves as the definitive evidence during regulatory inspections; verbal explanations are insufficient without supporting documentation. Ensuring a high-quality TMF from the outset is paramount for successful drug approval and regulatory compliance. * **Proactive Inspection Readiness:** Inspection readiness is not a reactive measure but an ongoing process that begins at the start of a study. A well-maintained TMF eliminates the need for last-minute scrambling to locate documents during an inspection. * **Risk-Based Assessment for QC:** Organizations should utilize risk-based assessment to strategically plan and execute quality control (QC) activities for their eTMF. This approach helps prioritize efforts on areas with the highest potential for quality issues and impact on study integrity. * **Definition of a Quality TMF:** A quality TMF is characterized by being complete, collected in a timely manner, and composed of high-quality records. It must tell the entire story of a clinical study independently, without requiring extensive additional explanation from staff. * **Shared Responsibility for TMF Content:** While clinical operations often manages the TMF, the responsibility for the quality of content extends to all functional areas contributing to the TMF, including data management, statistics, and others. Each contributor is accountable for the integrity of their submissions. * **TMF Reference Model:** The TMF Reference Model is an important industry standard that provides a standardized taxonomy and expected document types for the TMF, aiding in organization and completeness. The speaker has experience chairing revisions of this model. * **CFR Part 11 Compliance:** Experience with 21 CFR Part 11 is crucial for the validation of both clinical systems and TMF systems, ensuring the integrity, authenticity, and confidentiality of electronic records and signatures. * **Impact of Team Changes:** The TMF must be a standalone record set because study teams inevitably change over time. The documentation must be comprehensive enough to convey the study's narrative and compliance without relying on the institutional knowledge of individuals who may no longer be involved. * **Identifying High-Risk Artifacts:** Specific "artifacts" or types of documentation within the TMF are known to be significant risks for quality issues. Identifying and proactively addressing these high-risk areas is a critical component of effective TMF oversight. * **GCP and Data Integrity:** The TMF must provide evidence that the study was conducted in accordance with Good Clinical Practice (GCP) requirements and demonstrate the overall integrity of the study data. This is a core expectation of regulatory agencies. Key Concepts: * **eTMF (electronic Trial Master File):** A collection of essential documents that individually and collectively permit the evaluation of the conduct of a clinical trial and the quality of the data produced. In an electronic format, it's a regulated enterprise software system. * **TMF Quality Oversight:** The systematic process of ensuring that the TMF is complete, accurate, timely, and compliant with regulatory requirements and internal procedures. * **Risk-Based Approach:** A strategy for management and oversight that prioritizes activities based on the potential for quality issues to occur and the impact these deficiencies could have on the integrity of the TMF and overall GCP compliance. * **Good Clinical Practice (GCP):** An international ethical and scientific quality standard for designing, conducting, recording, and reporting trials that involve the participation of human subjects. * **21 CFR Part 11:** Regulations issued by the FDA that set forth the criteria under which electronic records and electronic signatures are considered trustworthy, reliable, and equivalent to paper records and handwritten signatures. * **Inspection Readiness:** The state of being fully prepared to present a complete, accurate, and compliant TMF to regulatory agencies during an inspection, demonstrating adherence to all relevant regulations and protocols. * **Functional Areas:** Different departments or groups within an organization (e.g., clinical operations, data management, statistics, medical affairs) that contribute to the conduct of a clinical trial and, consequently, to the content of the TMF. Tools/Resources Mentioned: * **TMF Reference Model:** An industry-standard, universally accepted taxonomy for the TMF, providing a standardized structure and naming convention for TMF documents. * **European Directive 2005:** Referenced as the source for the definition of what constitutes a Trial Master File.

TMF/eTMF Audit Strategies Trailer
Kathy Barnett
/@kathybarnett4070
Mar 2, 2016
This video provides an in-depth exploration of Trial Master File (TMF) and electronic TMF (eTMF) audit strategies, emphasizing their critical role in Good Clinical Practice (GCP) compliance and overall inspection readiness within clinical trials. The speaker, an industry consultant with 25 years of experience in pharma and clinical development, outlines practical approaches for conducting effective TMF audits. The presentation begins by setting the stage with objectives, including understanding the value of the TMF Reference Model, strategies for auditing both paper and eTMFs, and identifying artifacts that significantly impact quality and GCP compliance. A key focus is on leveraging the capabilities of an eTMF to enhance audit effectiveness and pinpoint potential inspection findings. The speaker highlights the significant evolution of TMF management over the last decade, with many organizations transitioning to eTMFs, while some still operate with paper or hybrid systems. A central theme is the TMF's role as the "first face to the regulator," underscoring that even the most successful clinical trial cannot lead to drug approval without comprehensive and compliant documentation. The discussion meticulously differentiates between audit and quality control (QC), defining audit as a process-focused evaluation that uses data to ensure adherence to established procedures, distinct from QC's data-driven oversight of specific information. This distinction is crucial for understanding the scope and objectives of a TMF audit. Furthermore, the video delves into the definition of a TMF, emphasizing its requirement to be a standalone set of documentation or records that can be understood by a regulator without extensive additional explanation. It must tell the complete story of a study, allowing an auditor to trace events and verify data integrity and GCP compliance. A critical insight shared is that the TMF is no longer solely a "clinical product" but rather a "total story" to which various functional areas, such as data management, biostatistics, and clinical trial materials, contribute significantly. The speaker's extensive background, including chairing the TMF Reference Model committee's ethics revisions and experience with 21 CFR Part 11 compliance for clinical and eTMF systems, lends substantial credibility and practical depth to the strategies presented. The strategies discussed aim to equip attendees with actionable insights for their day-to-day roles, whether in quality assurance or clinical operations. The TMF Reference Model is presented as a powerful tool for organizing audits efficiently and pinpointing GCP compliance issues. The speaker also touches upon critical files to review during an audit and methods for identifying trends in non-compliance, ensuring that the audit process is not just a checklist exercise but a strategic tool for maintaining high-quality trial conduct and regulatory adherence. Key Takeaways: * **TMF as the Regulator's First Impression:** The Trial Master File serves as the primary evidence for regulators regarding a trial's conduct. Without robust, compliant TMF documentation, even a successful trial cannot progress towards drug approval. * **Distinction Between Audit and QC:** Audits are process-focused, evaluating whether established procedures are followed, evidenced by data. Quality Control (QC) is data-driven, focusing on specific information oversight. Both are critical but serve different purposes. * **The TMF Reference Model's Value:** Utilizing the TMF Reference Model significantly enhances audit efficiency by providing a structured framework for organizing documentation and identifying potential GCP compliance gaps. * **Comprehensive TMF Definition:** A TMF must be a standalone, self-explanatory collection of records that tells the complete story of a clinical trial. It should allow an auditor or regulator to understand what happened, how it happened, and verify data integrity and GCP compliance without needing external explanations. * **TMF as a "Total Story" from All Functional Areas:** The TMF is no longer just a clinical department's responsibility. It aggregates documentation from all functional areas, including data management, biostatistics, and clinical trial materials, to provide a holistic view of the study. * **Leveraging eTMF for Enhanced Audits:** Electronic TMF (eTMF) systems offer powerful capabilities for identifying gaps and potential inspection findings more effectively than traditional paper-based systems, enabling more proactive compliance management. * **Importance of 21 CFR Part 11 Compliance:** The speaker's experience with 21 CFR Part 11 compliance for both clinical and eTMF systems highlights the critical need for electronic records and signatures to meet regulatory standards. * **Strategies for Identifying Non-Compliance Trends:** Effective audit strategies involve not just checking individual documents but also identifying critical files to review and spotting overarching trends in non-compliance that could indicate systemic issues. * **Impact on Drug to Patient Pathway:** The ultimate goal of a compliant TMF is to ensure that drugs can reach patients. Any deficiencies in TMF documentation can significantly impede regulatory approval processes. * **Practical Application of Audit Information:** The session aims to provide attendees with concrete strategies that can be directly applied in their daily roles within quality assurance or clinical operations to improve TMF management and audit readiness. Key Concepts: * **Trial Master File (TMF):** The essential collection of documents for a clinical trial that individually and collectively permit the evaluation of the conduct of a trial and the quality of the data produced. * **Electronic Trial Master File (eTMF):** A digital system for managing and storing TMF documents, offering enhanced searchability, audit trails, and compliance features. * **Good Clinical Practice (GCP):** An international ethical and scientific quality standard for designing, conducting, recording, and reporting trials that involve the participation of human subjects. * **TMF Reference Model:** A standardized, hierarchical model for organizing TMF documents, promoting consistency and efficiency across trials and organizations. * **21 CFR Part 11:** Regulations issued by the FDA that set forth the criteria under which electronic records and electronic signatures are considered trustworthy, reliable, and equivalent to paper records and handwritten signatures. * **Inspection Readiness:** The state of being prepared for regulatory inspections, ensuring all documentation and processes are compliant and readily accessible. * **Audit vs. Quality Control (QC):** Audit focuses on evaluating processes and adherence to them, using data as evidence. QC focuses on specific data points and information oversight. Tools/Resources Mentioned: * **TMF Reference Model:** A key framework for organizing and auditing TMFs. * **eTMF Systems:** Electronic platforms designed for managing Trial Master Files, offering capabilities for improved audit effectiveness.

Veeva Systems is Bringing Healthcare Data Into the 21st Century
The Motley Fool
/@MotleyFool
Feb 15, 2016
This discussion provides an analysis of Veeva Systems, positioning the company as a critical player in bringing healthcare data management into the 21st century. The speakers emphasize Veeva's significant first-mover advantage within the life sciences data management sector. The core of Veeva's business is segmented into two major components: the foundational Customer Relationship Management (CRM) business and the specialized content and data management system, Veeva Vault. The analysis establishes that Veeva's target market is the largest players in the industry, noting that 34 of the 50 largest pharmaceutical companies utilize their services, including giants like Pfizer and Merck. The conversation delves into the function and necessity of Veeva Vault, which is described as a system designed to handle the immense volume of information generated throughout the drug development lifecycle. This includes the complex data associated with clinical trials, regulatory approval records, and related documentation. The primary value proposition of Vault is its ability to securely store, share, and manage these master files, facilitating necessary transfers to regulatory parties such as the FDA. The speakers underscore the sheer scale of the data challenge in pharmaceuticals, noting that the process of bringing a single drug to market is a "mind-boggling task" that typically lasts over a decade and costs an estimated $2 billion per drug. The analysis highlights that the pharmaceutical industry's need for robust, secure, and efficient data management is driven by the high cost and duration of the drug approval process. Given the tremendous amount of information generated, the industry requires a specialized solution for storage and accessibility. The speakers conclude that cloud-based software, which Veeva provides, is an "intriguing way of meeting this need," signaling the shift away from legacy systems toward modern, scalable, and secure cloud infrastructure for mission-critical life sciences data. ### Detailed Key Takeaways * **Veeva Systems' Dual Business Focus:** Veeva operates through two primary divisions critical to the life sciences sector: the core CRM business, focused on customer relationship management (essential for commercial operations), and Veeva Vault, a specialized management system for regulated content and data. * **Dominant Market Penetration:** Veeva holds a commanding position within the pharmaceutical ecosystem, serving 34 of the 50 largest pharmaceutical companies, including major players like Pfizer and Merck, indicating its status as an industry standard for enterprise-level data solutions. * **Veeva Vault's Regulatory Significance:** Vault serves as the central repository for highly sensitive and regulated data, including information from clinical trials and drug approval records. Its function is to securely store, share, and enable the transfer of these "master files" to regulatory bodies such as the FDA, which is critical for compliance and audit readiness. * **Addressing the Scale of Pharmaceutical Data:** The drug development process generates a "tremendous amount of information" due to its complexity, which can last over a decade and cost approximately $2 billion per drug. This massive data volume necessitates specialized, secure, and scalable data management solutions like those offered by Veeva. * **First-Mover Advantage in Cloud Solutions:** Veeva capitalized on being an early adopter and provider of cloud-based software tailored specifically for the life sciences industry, offering a modern, efficient, and secure alternative to traditional data storage and management methods. * **Strategic Importance for Commercial Operations:** While Veeva Vault focuses on R&D and regulatory data, the core Veeva CRM platform is indispensable for commercial operations, sales force effectiveness, and managing interactions with healthcare professionals (HCPs)—a key area for AI-driven optimization and sales operations assistance. * **Implications for Data Engineering:** The requirement to manage, secure, and transfer master files to the FDA underscores the need for robust data engineering and pipeline development to ensure data integrity, auditability, and compliance (specifically GxP and 21 CFR Part 11 standards). * **Opportunity for AI Integration:** The vast, structured datasets managed within Veeva Vault and CRM represent an ideal foundation for implementing AI and LLM solutions. These systems can be leveraged for intelligent automation, predictive analytics on clinical trial data, and enhancing commercial intelligence through tools like generative AI sales assistants. * **High Barrier to Entry:** The complexity, regulatory requirements, and massive data volume associated with drug development create a high barrier to entry for competitors, reinforcing Veeva's market position and the necessity for specialized consulting expertise to navigate these systems. ### Tools/Resources Mentioned * **Veeva Systems:** The primary life sciences data management company discussed. * **Veeva CRM:** The core customer relationship management platform for pharmaceutical sales and commercial operations. * **Veeva Vault:** The content and data management system used for clinical trials, regulatory submissions, and master file storage. * **FDA (Food and Drug Administration):** The key regulatory party receiving master files and approval records managed through Vault. ### Key Concepts * **Life Sciences Data Management:** The specialized field of handling, securing, and managing the massive, complex, and highly regulated data generated by pharmaceutical and biotech companies, particularly during R&D and commercialization. * **Clinical Trials and Approval Records:** The extensive documentation and data generated during the multi-phase testing required to prove a drug's safety and efficacy, which must be securely stored and accessible for regulatory review. * **Master Files:** Official, comprehensive sets of documents and data related to a drug's development and manufacturing process that are submitted to regulatory authorities. * **Cloud-Based Software:** Utilizing internet-based servers for data storage and management, offering scalability, security, and accessibility crucial for global pharmaceutical operations, contrasting with older, on-premise systems.

Just How Big is Veeva Systems’ Addressable Market?
The Motley Fool
/@MotleyFool
Feb 15, 2016
This video provides a focused financial and strategic analysis of Veeva Systems, specifically examining the size and growth potential of its addressable market as of early 2016. The discussion centers on determining the total market opportunity for Veeva, a critical factor for investors evaluating the company as a potential "Rule Breaker" investment—a term used by The Motley Fool to describe high-growth, early-stage companies with massive potential, often before achieving full profitability. The speakers, Sean O’Reilly and Kristine Harjes, break down Veeva’s market opportunity into its two primary business segments: the established CRM platform and the rapidly growing Vault suite. The analysis highlights that while Veeva’s CRM platform—the company’s initial success and primary revenue driver at the time—was estimated to hold a $2 billion market opportunity, the newer Vault software suite was projected to match that size, representing another $2 billion market. This projection was based on estimates from Veeva’s CEO, signaling that Vault was positioned as the primary growth engine moving forward, despite only contributing 25% of the company’s total revenue at the time of the recording. The conversation establishes that the combined market size of these two core platforms is approximately $4 billion, underscoring the substantial runway for growth within the pharmaceutical and life sciences industries. Furthermore, the speakers extrapolate the total addressable market (TAM) for Veeva Systems beyond the two main platforms. They account for a third, very young business segment that was not yet substantial enough to warrant detailed discussion but was estimated to contribute an additional $1 billion to the overall TAM. This led to a final, estimated total addressable market of $5 billion for Veeva Systems. This comprehensive market sizing exercise provides a strategic perspective on the company’s future trajectory, emphasizing that its value proposition—bringing healthcare data into the 21st century—was addressing a massive, underserved need within the highly regulated life sciences sector. Key Takeaways: * **Veeva’s Strategic Growth Driver:** The Vault software suite was identified as the key growth engine for Veeva Systems, despite contributing only 25% of the company's revenue at the time of the analysis. This indicates a strategic shift toward content and data management solutions beyond traditional CRM. * **Equal Market Opportunity for Vault and CRM:** The CEO of Veeva estimated that the Vault platform had an addressable market size equivalent to the established CRM platform, with both segments independently representing a $2 billion market opportunity. * **Massive Total Addressable Market (TAM):** The total estimated addressable market for Veeva Systems was pegged at approximately $5 billion, derived from $2 billion for CRM, $2 billion for Vault, and an additional $1 billion estimated for emerging, nascent business lines. * **Focus on Regulated Content Management:** The rapid growth potential attributed to Vault underscores the pharmaceutical industry's critical need for modern, compliant, cloud-based solutions for managing regulated content across R&D, clinical trials, quality, and commercial operations. * **Implications for Life Sciences Consultants:** Consulting firms specializing in the life sciences sector, like IntuitionLabs.ai, must recognize that the growth opportunity lies equally in the Vault ecosystem (spanning clinical, regulatory, quality, and medical affairs) as it does in the traditional Commercial CRM space. * **Investment in Emerging Segments:** The inclusion of a $1 billion estimate for a "very young" third business segment suggests Veeva’s continuous strategy of identifying and penetrating new, related areas within the life sciences value chain, which could involve data analytics, AI, or specialized vertical applications. * **Understanding Veeva’s Dominance:** The sheer size of the TAM discussed confirms Veeva’s position as the dominant, industry-specific cloud provider, making expertise in their platforms essential for any technology provider serving the pharmaceutical sector. * **Prioritizing Vault Expertise:** For firms offering AI and data engineering services, focusing development efforts on integrating with or enhancing the Vault platform—which manages core regulated data—is crucial for accessing the largest future growth areas within the Veeva ecosystem. Key Concepts: * **Addressable Market (TAM):** The total potential revenue opportunity available within a defined market segment for a specific product or service. The video uses this metric to gauge Veeva's long-term growth potential. * **Veeva Vault:** A suite of cloud-based applications designed specifically for the life sciences industry to manage content and data across various functions, including clinical, regulatory, quality, and commercial. * **Veeva CRM:** The cloud-based customer relationship management platform tailored for pharmaceutical sales and marketing teams, serving as Veeva’s foundational product. * **Rule Breaker Investment:** A term used by The Motley Fool to describe companies with disruptive technology, strong leadership, and a massive addressable market, suggesting potential for exponential growth.

eTMF Connect Demo | Document Authoring and Collaboration
Montrium
/@Montrium
Feb 9, 2016
This video provides an in-depth demonstration of the eTMF Connect solution, focusing specifically on the lifecycle of document authoring, collaboration, and filing within a regulated clinical trial environment. The demonstration follows the process from the perspective of David, a clinical author, who needs to upload and finalize a new study protocol document. The core objective of the system is to simplify the population of the Trial Master File (TMF) by centrally managing documentation within a pre-defined, compliant structure, ensuring that content is created, reviewed, and filed correctly from the outset. The demonstration highlights the system's reliance on industry standards to enforce structure and compliance. eTMF Connect organizes clinical records using virtual cabinet drawers and process zones that are pre-defined and strictly aligned with both the DIA TMF Reference Model and the DIA EDM Reference Model. This structural alignment ensures that users are guided to the exact location where specific documents must reside. When David initiates the creation of a new protocol, the system presents a list of available document types for that specific section. Crucially, the system utilizes pre-defined templates, indicated by a Microsoft Word icon, which guarantees that the author begins with the correct format and required boilerplate content, mitigating errors and ensuring consistency across studies. A key feature demonstrated is the seamless integration with Microsoft Word for authoring and collaboration. Upon selecting the protocol document type, the template opens directly in Word, with essential document properties and metadata pre-populated in a dedicated tab. Once the initial editing is complete, the author can immediately define the subsequent workflow steps, including specifying reviewers and approvers directly within the metadata bar, and assigning themselves as the document owner. When the document is checked back into the system, eTMF Connect automatically assigns a unique name and number, files it in the correct TMF location, and updates its status to "Draft." The system excels in managing the collaborative review process. Once the document is checked in, all specified reviewers receive automated tasks, initiating the collaborative editing phase. The platform supports simultaneous tracking of edits, commenting, and acceptance/rejection of changes, all managed through the familiar Microsoft Word interface. This centralized feedback mechanism allows the document owner to consolidate all reviewer input into a single file, preparing the document for the final approval workflow. This streamlined process ensures efficiency and maintains a complete audit trail of all changes made during the critical drafting and review stages of clinical documentation. Key Takeaways: • **Regulatory Structure Enforcement:** The eTMF system enforces compliance by aligning its document process zones and filing structure directly with the DIA TMF Reference Model and DIA EDM Reference Model, ensuring documents are categorized and stored correctly for regulatory inspection readiness. • **Template-Driven Consistency:** Utilizing pre-defined document templates (accessible via a Word integration) ensures that clinical authors start with the correct format and required content, significantly reducing the risk of non-compliant or incomplete documentation. • **Metadata Automation:** Document properties and metadata are pre-populated when a document is created from a template, automating the data entry process and ensuring accuracy before the author begins editing the core content. • **Integrated Collaborative Editing:** The platform facilitates real-time, simultaneous collaboration within the native Microsoft Word environment, allowing reviewers to track edits, leave comments, and manage changes efficiently without needing to export or re-upload files. • **Automated Workflow Routing:** Authors can define the entire review and approval workflow (specifying reviewers and approvers) directly within the document’s metadata, triggering automated task assignments upon check-in. • **Unique Identifier Assignment:** Upon saving or checking in a document, the system automatically assigns a unique name and number, standardizing nomenclature and simplifying document retrieval and tracking throughout the trial lifecycle. • **Centralized Document Status Tracking:** The system maintains clear visibility into the document lifecycle by automatically updating the status (e.g., "Draft") based on the current stage of the workflow, providing immediate context to all users accessing the TMF section. • **Efficiency in Clinical Operations:** By automating template generation, metadata entry, filing location, and workflow initiation, the system significantly reduces the manual effort and potential for human error associated with managing critical clinical documentation. Key Concepts: * **eTMF (Electronic Trial Master File):** A system used by life sciences companies to manage, store, and track essential clinical trial documents required for regulatory compliance. * **DIA TMF Reference Model:** A standardized, hierarchical structure for organizing the TMF, widely adopted in the industry to ensure consistency and inspection readiness. * **DIA EDM Reference Model:** The Drug Information Association's Electronic Document Management Reference Model, providing guidance on managing electronic regulatory submissions and documentation. * **Document Authoring and Collaboration:** The process of creating, editing, reviewing, and approving documents, often involving simultaneous input from multiple team members while maintaining an auditable change history. Tools/Resources Mentioned: * **eTMF Connect:** The specific eTMF solution demonstrated. * **Microsoft Word:** Used as the integrated authoring and collaborative editing tool. * **DIA TMF Reference Model:** The foundational standard used for structuring the TMF within the system.
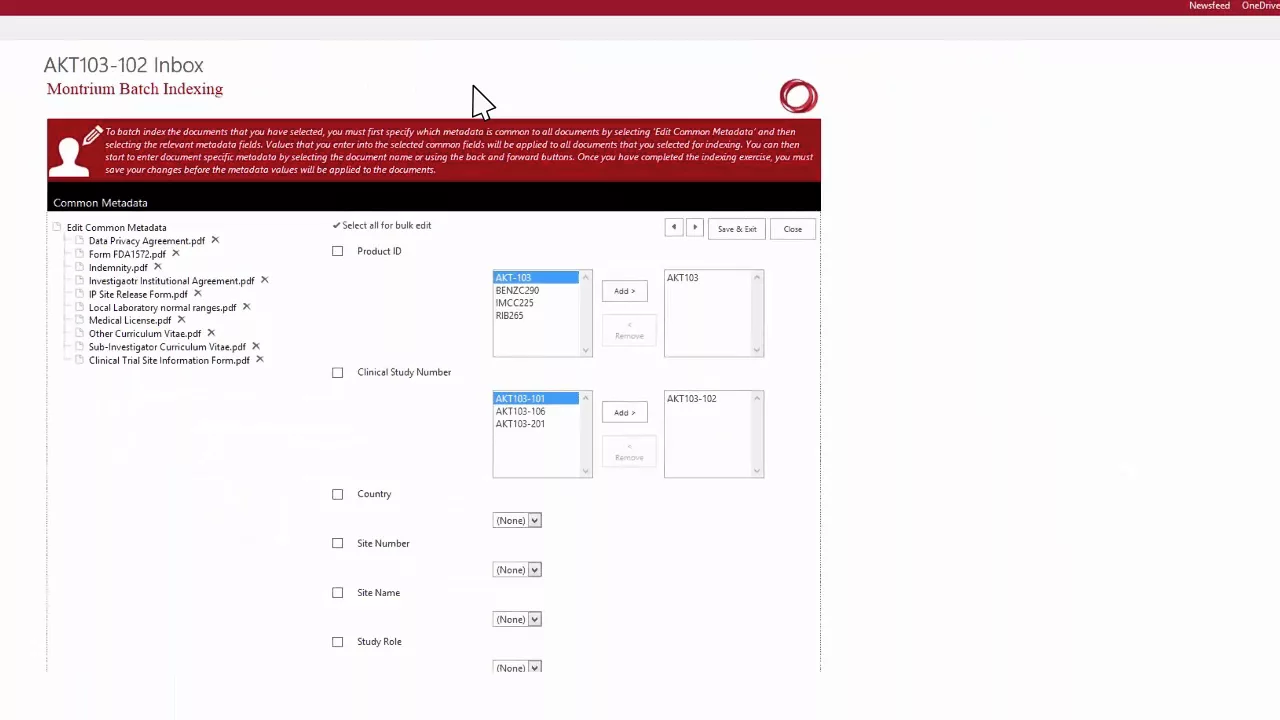
eTMF Connect Demo | Batch Indexing and Uploading Documents
Montrium
/@Montrium
Feb 9, 2016
This video provides a demonstration of the bulk upload and batch indexing capabilities within the eTMF Connect solution, focusing on streamlining the process of ingesting clinical trial documentation. The context is set within the "clinical inbox," a designated entry point for documentation sent to specific study and site email inboxes. The core objective of the workflow is to efficiently classify, index, and automatically route large volumes of documents to their correct locations within the Trial Master File (TMF), ensuring compliance and audit readiness. The demonstration highlights how the system minimizes manual effort and the risk of human error associated with repetitive metadata entry. The workflow begins with a user, "David," navigating to the clinical inbox for his study. He initiates the bulk upload by dragging and dropping multiple files from his desktop into the working area. Once the upload is complete, the critical step of classification begins. Using the batch indexing feature, David selects all newly uploaded documents and activates an indexing wizard. This wizard is designed to handle common metadata fields across all selected documents simultaneously, a significant time-saver. For instance, if all documents pertain to the same study or site, that information is entered once and applied universally. A key feature demonstrated is the ability to lock common metadata while allowing for document-specific classification. The presenter illustrates that while general metadata (like study ID) is locked, David can navigate through each document individually within the wizard to input unique details, such as specific approval dates or version numbers. Once the indexing is finalized, the system leverages a central component called the "file plan." The file plan is described as a comprehensive, industry-standard list of all required document types and content types within the TMF. The system uses the newly classified metadata to consult the file plan, automatically determining the correct routing and filing location for each document. This automated routing ensures that documents are placed according to regulatory and organizational standards, moving them out of the temporary clinical inbox and into the permanent, structured TMF library. Key Takeaways: • **Efficiency in Clinical Document Ingestion:** The primary value proposition of the batch indexing feature is the dramatic reduction in time required to process incoming clinical documentation, moving away from single-document uploads and indexing toward scalable bulk processing. • **Minimizing Repetitive Data Entry:** The batch indexing wizard allows users to apply common metadata (e.g., study ID, site number) across multiple documents simultaneously, eliminating the need to repeatedly input identical information and reducing the likelihood of transcription errors. • **Structured Metadata Classification:** The system supports a hybrid indexing approach where common metadata can be locked, ensuring consistency, while allowing for granular, document-specific metadata (like approval dates or version numbers) to be added individually within the same workflow. • **Automated Routing via File Plan:** The system relies on a central, industry-standard "file plan" to automatically route documents based on the metadata classified during the indexing process. This ensures that documents are filed in the correct TMF structure, critical for GxP compliance and audit readiness. • **Importance of the Clinical Inbox:** The clinical inbox serves as a critical staging area and entry point for external documentation, often received via study or site-specific email addresses, providing a controlled environment before documents are formally committed to the TMF. • **Compliance and Standardization:** By utilizing a standardized file plan and enforcing metadata classification before filing, the system helps life sciences organizations maintain a compliant and standardized TMF structure, which is essential for regulatory inspections (e.g., FDA, EMA). • **Relevance to AI Integration:** The highly structured nature of this eTMF workflow (metadata-driven classification and routing) creates an ideal environment for integrating AI/LLM solutions. IntuitionLabs.ai could leverage this structure to build AI agents that automatically extract metadata from incoming documents in the clinical inbox, pre-populating the indexing wizard and further automating the classification process. • **Data Engineering Foundation:** The reliance on a robust "file plan" and structured metadata emphasizes the need for strong data engineering practices to maintain the integrity of the TMF structure and ensure accurate routing and retrieval of clinical data. Tools/Resources Mentioned: * eTMF Connect (Montrium) * Clinical Inbox * Indexing Wizard * File Plan (Central list of document types/content types) Key Concepts: * **eTMF (Electronic Trial Master File):** A system used by sponsors and CROs to manage essential clinical trial documentation in a compliant, centralized, and standardized manner. * **Batch Indexing:** The process of classifying and applying metadata to multiple documents simultaneously, significantly speeding up the documentation workflow. * **Clinical Inbox:** A temporary holding area or staging environment where incoming clinical documents are collected, reviewed, and prepared for formal classification and filing into the TMF. * **File Plan:** A predefined, hierarchical structure based on industry standards (like the TMF Reference Model) that dictates where specific document types must be filed within the eTMF system for regulatory compliance.
eTMF Connect Demo | Accelerating Clinical Study and Site Start up
Montrium
/@Montrium
Feb 9, 2016
This video provides a demonstration of eTMF Connect, a solution designed to accelerate the clinical study and site startup process by leveraging pre-existing trial information and automating traditionally manual tasks. The core purpose of the system is to centralize and standardize clinical records, allowing both sponsors and Contract Research Organizations (CROs) to efficiently manage and access critical documentation in real-time. The demonstration focuses on the study management feature, highlighting how users like "David" can organize, manage, and create new studies and sites with minimal effort, effectively eliminating the cumbersome paper-based processes often associated with clinical trial initiation. The demonstration walks through the user experience, beginning with navigating to the study management area where a full overview of current studies, sites, and staff is available. The system utilizes accordion-style drop-downs to provide quick access to high-level study and site information. A key feature showcased is the creation of a new site using a dynamic form. This form incorporates "intelligent lookups" and dynamic elements that connect the entire study structure. As the user selects fields in drop-down menus (e.g., selecting the study), associated fields are automatically populated with relevant options, significantly reducing the potential for human error and ensuring data consistency across the trial setup. This automation is central to the system's value proposition of accelerating startup phases. Once the site creation form is completed and submitted, the site startup is immediately initiated. The system then transitions seamlessly into site-level document management. The video highlights three distinct methods for document ingestion, catering to different user workflows: uploading documents one at a time with individual classification, bulk uploading via drag-and-drop functionality from a desktop folder, or sending documents via email to an inbox for later classification. This flexibility in document handling ensures that study teams can quickly and compliantly populate the Electronic Trial Master File (eTMF) with essential site documents, further streamlining the transition from site setup to operational readiness. The underlying methodology of eTMF Connect emphasizes standardization and centralization. By providing a single source of truth for clinical records, the solution ensures that all stakeholders—from sponsors to CROs—are working with the most current and compliant documentation. The focus on automating the initial setup phase, including the creation of sites and the initial population of the TMF, directly addresses a major bottleneck in clinical trials: the time-consuming and error-prone administrative burden of study initiation. This approach is critical for maintaining GxP compliance and ensuring audit readiness from the earliest stages of the clinical lifecycle. Key Takeaways: • **Automation of Study/Site Startup:** The eTMF Connect solution leverages pre-existing clinical trial information to automate the creation and setup of new studies and sites, significantly accelerating the traditionally slow and manual study initiation phases. • **Intelligent Data Consistency:** The system utilizes dynamic forms with "intelligent lookups" where selecting one field automatically populates associated fields with relevant options, drastically minimizing data entry errors and ensuring high data quality across the clinical directory. • **Centralized Clinical Records Management:** The platform serves as a central hub for clinical records, enabling both sponsors and CROs to contribute, manage, and access essential clinical documents in real-time, which is crucial for collaborative trials. • **Elimination of Paper Processes:** The core benefit highlighted is the effective elimination of paper-based processes during study and site startup, moving all critical documentation and management tasks into a digital, compliant environment. • **Flexible Document Ingestion:** The system supports multiple methods for populating the eTMF with site-level documents, including single document uploads with classification, bulk drag-and-drop uploads, and email-to-inbox functionality for later classification, accommodating diverse user preferences. • **Structured Information Management:** The study management area provides a comprehensive overview of studies, sites, and staff, utilizing an accordion-style interface for quick access to high-level information, improving organizational efficiency for study teams. • **Regulatory Compliance Foundation:** As an eTMF vendor with over 10 years of experience, the solution inherently supports the necessary structure and audit trails required for GxP and 21 CFR Part 11 compliance, ensuring that clinical documentation is always inspection-ready. • **Focus on User Workflow:** The demonstration emphasizes a user-centric approach, showing how a user ("David") can quickly navigate, create, and manage complex clinical information through intuitive quicklinks and streamlined forms. Tools/Resources Mentioned: * eTMF Connect (Montrium) Key Concepts: * **eTMF (Electronic Trial Master File):** A system used by life sciences companies to manage, store, and archive the essential documents of a clinical trial in a digital format, ensuring regulatory compliance and audit readiness. * **Clinical Study and Site Start-up:** The initial phase of a clinical trial involving regulatory approvals, site selection, contract negotiation, and the collection of essential documents before patient enrollment can begin. * **Intelligent Lookups:** A feature in data entry forms where the system uses existing data relationships to suggest or automatically populate related fields, reducing manual input and improving data accuracy. * **Sponsors and CROs (Contract Research Organizations):** The primary entities involved in clinical trials; sponsors fund the research, and CROs often manage the execution of the trial, requiring shared access to the eTMF.
eTMF Connect Demo | Revising Clinical Documentation
Montrium
/@Montrium
Feb 9, 2016
This video demonstrates the compliant electronic workflow for revising clinical documentation within eTMF Connect, an Electronic Trial Master File (eTMF) solution tailored for the life sciences sector. The demonstration establishes the critical need for a controlled revision process, acknowledging that clinical studies and organizational requirements frequently necessitate changes to existing documentation. The platform is positioned as a tool that centralizes and standardizes clinical records, facilitating real-time access and contribution for both sponsors and Contract Research Organizations (CROs) while maintaining regulatory integrity. The core of the demonstration focuses on the user experience of initiating a change. The process begins with the authorized user navigating to the document library and utilizing a context menu to trigger a dynamic revision request form. A key efficiency feature highlighted is the system's ability to automatically pre-populate the form with existing metadata from the document being revised, minimizing manual entry and ensuring accuracy. The user is then required to formally document the rationale for the revision, specify the type of change, and designate a revision request approver, formalizing the justification for the change before any physical editing occurs. Once the revision request is submitted, the system initiates a dedicated workflow. This workflow’s primary purpose is to manage the transition of the document from a controlled, read-only state (typically PDF) to an editable format (such as Word). This controlled unlocking mechanism is essential for maintaining GxP compliance, ensuring that unauthorized changes cannot be made. Upon successful completion of this initial approval workflow, the original word document is unlocked, allowing the user to make the necessary content edits. The final step demonstrated is the initiation of the subsequent, separate Review and Approval workflow, which validates the revised content before the document is finalized, re-controlled, and archived within the eTMF system, ensuring a complete and auditable history of the document's lifecycle. Key Takeaways: * **Mandate for Electronic Change Control:** The necessity for revising clinical documentation due to evolving study protocols or organizational shifts mandates the use of a robust, electronic change control system to ensure all modifications are tracked, justified, and compliant with regulatory standards. * **Streamlined Initiation via Dynamic Forms:** The platform utilizes dynamic revision request forms that capture essential metadata (rationale, revision type) upfront, formalizing the change control process immediately and providing the necessary justification for audit trails. * **Metadata-Driven Efficiency:** The automatic pre-population of revision request forms with existing document metadata significantly enhances data accuracy and operational efficiency by reducing the need for manual transcription of document details. * **Controlled Document State Transition:** The system enforces a critical control point by managing the transition of documents from a controlled, read-only state (e.g., PDF) to an editable format (e.g., Word) only after the formal Revision Request Workflow has been approved. * **Segregation of Approval Workflows:** The revision process is structured into two distinct, auditable workflows: the initial Revision Request Approval (authorizing the edit) and the subsequent Content Review and Approval (validating the changes), ensuring clear separation of duties and comprehensive tracking. * **Importance of Approver Designation:** Users must designate a specific revision request approver, establishing accountability and ensuring that the decision to unlock and modify the clinical document is formally documented and authorized before editing commences. * **Support for Sponsor/CRO Collaboration:** The eTMF solution is designed to centralize and standardize clinical records, enabling seamless and compliant contribution and access for both the trial sponsors and their contracted CROs in real-time. * **Auditability and Compliance Foundation:** The entire electronic process—from the initial request to the final saving of the revised document—is engineered to maintain a complete audit trail, satisfying stringent regulatory requirements related to document control and data integrity (critical for GxP and 21 CFR Part 11 adherence). Tools/Resources Mentioned: * **eTMF Connect:** A specialized Electronic Trial Master File (eTMF) solution for life sciences companies, provided by Montrium. Key Concepts: * **eTMF (Electronic Trial Master File):** The digital repository for all essential documents related to a clinical trial, required for regulatory compliance and reconstruction of the trial history. * **Revision Request Rationale:** The required justification provided by the user explaining why a change to a controlled clinical document is necessary, forming a key component of the audit trail. * **Workflow Management:** The automated routing and tracking of documents through predefined steps (editing, review, approval) to ensure compliance, consistency, and timely completion of regulated tasks.

eTMF Connect Demo | Introduction
Montrium
/@Montrium
Feb 9, 2016
This video provides a brief introductory demonstration of eTMF Connect, the electronic Trial Master File solution developed by Montrium. The primary purpose of the solution is to help life sciences organizations, including pharmaceutical sponsors and Contract Research Organizations (CROs), centralize, standardize, and maintain critical documentation related to clinical trials. The presentation establishes eTMF Connect as a compliant document management system designed to streamline the complex process of managing clinical records across multiple geographical locations and participating entities. The solution is positioned as a secure, web-browser accessible platform, eliminating the need for specific software downloads and allowing for immediate, real-time access and contribution from various stakeholders. Key users include sponsors, CROs, and investigated sites, all of whom require a unified environment for planning, collecting, and maintaining documentation. By centralizing these records, eTMF Connect aims to overcome the logistical and compliance challenges inherent in decentralized clinical trial documentation, promoting efficiency and data integrity throughout the trial lifecycle. A core feature highlighted is the solution’s alignment with the Drug Information Association (DIA) Trial Master File Reference Model. This alignment is critical, as the DIA model provides an industry-standard structure for organizing TMF content, ensuring consistency and completeness. Leveraging this standardized technology stack is presented as a mechanism to promote "iron site compliance," seamless collaboration between disparate teams, and overall increased productivity in clinical operations. The introduction concludes by setting the stage for a detailed demonstration of the solution’s capabilities, emphasizing its role as a leading, compliant eTMF vendor with over a decade of experience in the life sciences sector. Key Takeaways: • **Centralized Clinical Documentation Management:** The eTMF Connect solution provides a single, unified environment for managing all clinical trial documentation, which is essential for ensuring data consistency and accessibility for sponsors, CROs, and investigative sites globally. • **Real-Time Collaboration for Regulated Data:** The platform enables sponsors and CROs to contribute and access important clinical documents in real time, facilitating seamless collaboration across organizational boundaries while maintaining strict control over regulated content. • **Regulatory Alignment via DIA Reference Model:** The system is explicitly aligned with the DIA Trial Master File Reference Model, which is a crucial factor for organizations seeking to standardize their TMF structure and ensure that documentation meets industry best practices for completeness and quality. • **Focus on Site Compliance and Audit Readiness:** By promoting a standardized technology stack and centralized repository, the eTMF solution is designed to enhance "iron site compliance," meaning the documentation is organized and maintained in a manner that is consistently ready for regulatory audits and inspections. • **Accessibility and Deployment Flexibility:** eTMF Connect is accessible securely through a standard web browser, eliminating the need for specific software installations. It offers flexible deployment options, being available both on-premise and in the cloud, catering to different organizational IT infrastructure preferences. • **Streamlining Clinical Operations:** The core value proposition is increased productivity achieved by simplifying the processes of planning, collecting, and maintaining clinical trial documentation, thereby reducing administrative burden on clinical operations teams. • **Targeting the Life Sciences Ecosystem:** The solution directly addresses the needs of key players in the life sciences industry, specifically pharmaceutical sponsors and Contract Research Organizations (CROs), who are responsible for the execution and regulatory oversight of clinical trials. • **Leveraging Vendor Expertise:** Montrium positions itself as a leading eTMF vendor with over 10 years of experience, suggesting a mature, proven solution that has evolved to meet complex regulatory and operational demands in document management. Tools/Resources Mentioned: * **eTMF Connect:** The electronic Trial Master File software solution provided by Montrium. Key Concepts: * **eTMF (Electronic Trial Master File):** A digital system used to store, manage, and track all essential documents required to reconstruct and evaluate a clinical trial. It is a critical component of regulatory compliance (e.g., FDA, EMA) in clinical operations. * **Sponsors and CROs (Contract Research Organizations):** The primary entities responsible for initiating, managing, and overseeing clinical trials. The eTMF system is designed to facilitate document sharing and collaboration between these two parties. * **DIA Trial Master File Reference Model:** A standardized, hierarchical structure developed by the Drug Information Association (DIA) used by the industry to organize the content of the TMF. Adherence to this model ensures consistency, quality, and ease of review for regulatory bodies. * **Iron Site Compliance:** A term used to describe the state of having complete, accurate, and readily accessible documentation at the investigative site level, ensuring compliance with regulatory requirements and minimizing findings during audits.

Veeva CEO Peter Gassner on Managing a Growing Industry Cloud Company
Veeva Systems Inc
@VeevaSystems
Dec 11, 2015
This video features Veeva Co-founder and CEO Peter Gassner discussing the critical operational and cultural challenges involved in scaling an industry cloud company focused on the pharmaceutical and life sciences sectors, specifically targeting the transition from a $100 million revenue company toward the $1 billion milestone. Gassner frames the journey not as a strategic puzzle, but as a demanding exercise in execution, emphasizing that success requires constant organizational reinvention and an unwavering commitment to customer success. A primary focus of the discussion is the necessity of scaling execution and processes. Gassner highlights that the operational frameworks and leadership styles that succeed at a $100 million valuation become liabilities at $1 billion. He provides a specific example related to customer relationship management: a company accustomed to managing numerous $1 million annual relationships must completely overhaul its approach when it begins handling critical $50 million annual relationships with major customers. This shift necessitates a fundamental change in process, which is inherently difficult because organizational inertia makes employees comfortable with existing routines. Gassner stresses the need for leaders to actively stop and ask, "What should I be doing?" rather than simply continuing "What am I doing?" to break this cycle of complacency. Beyond operational scaling, Gassner dedicates significant attention to the cultural imperative of customer success and the mitigation of organizational arrogance. He asserts that authentic care for the customer must originate with the CEO and genuinely permeate the entire management team and employee base, noting that this commitment cannot be faked. Crucially, he addresses the unique danger faced by market leaders. Given Veeva’s trajectory toward achieving a "very high percentage market center in pharma CRM" (referencing their foundational product), the natural gravitational pull is toward arrogance. Gassner warns that customers instinctively rebel against vendor arrogance, making it a severe threat to long-term viability. He identifies arrogance as the company’s "boogeyman," and the only effective defense is maintaining constant awareness of this danger and proactively communicating the need for humility and partnership throughout the organization. Key Takeaways: • **Scaling Execution is the Hardest Part:** Achieving significant growth (e.g., $100M to $1B) is primarily an execution challenge, not just a strategic one. IntuitionLabs.ai must focus on building robust, scalable delivery models for its AI and consulting services that can withstand exponential client growth and complexity. • **Process Reinvention is Mandatory for Growth:** Operational processes that worked at a smaller scale, such as leadership structures and relationship management, must be constantly reinvented and upgraded to support larger revenue targets and more complex client needs. • **Evolving Customer Relationship Management:** As IntuitionLabs.ai secures larger contracts with major pharmaceutical clients, the relationship management approach must shift from generalized account handling to specialized, high-touch engagement tailored for multi-million dollar partnerships. • **Authentic Customer Care Must Be Top-Down:** Genuine commitment to customer success must be an authentic cultural value driven by the leadership team, as this trust is critical for long-term partnerships within the highly regulated life sciences environment. • **Beware the Gravity of Arrogance:** Market success and platform dominance (like that of Veeva CRM) naturally lead to organizational arrogance, which is highly detrimental to customer relationships. IntuitionLabs.ai must proactively foster a culture of humility and service to counteract this tendency, especially when consulting on proprietary, dominant platforms. • **Arrogance is the "Boogeyman":** Leaders must identify arrogance as a primary cultural threat and continuously educate their teams about the importance of maintaining a respectful, service-oriented posture, regardless of the company's or the platform's market share. • **Overcoming Organizational Inertia:** It is difficult to change established processes because people become comfortable with what they are doing. Leaders must force self-reflection by asking, "What should we be doing?" rather than simply continuing "What are we doing?" to ensure necessary scaling changes are implemented. • **Validation of Industry Cloud Focus:** The CEO's focus on building a deep, specialized industry cloud for pharma and life sciences validates IntuitionLabs.ai’s strategy of combining AI expertise with deep vertical knowledge of the pharmaceutical ecosystem. Key Concepts: * **Industry Cloud:** A software model focused entirely on serving the specific needs of a single vertical industry (in this case, pharmaceutical and life sciences), allowing for deep specialization and compliance integration. * **Execution Scaling:** The process of upgrading internal operational processes, leadership capacity, and workflows to support massive increases in revenue and organizational size, moving beyond the capabilities of a startup or mid-sized company. * **Organizational Arrogance:** The cultural pitfall where a successful or market-dominant company begins to prioritize its own needs or perceived superiority over genuine customer partnership, leading to customer dissatisfaction and rebellion.

Veeva CEO Peter Gassner Shares Veeva's Story
Veeva Systems Inc
/@VeevaSystems
Dec 10, 2015
This presentation features Veeva co-founder and CEO Peter Gassner sharing the strategic journey and foundational principles that guided Veeva Systems to become a leader in the industry cloud for the pharmaceutical and life sciences sectors. Delivered at the Emergence Capital Industry Cloud Forum, the talk provides a retrospective on the company's growth, which, at the time of the recording, included a $400 million annual run rate, 1,400 employees, and mid-20s net margins, with revenue and staff split evenly between the US and international markets. Gassner emphasizes that Veeva’s core mission is defined by its vision—building the industry cloud—and its three core values: Customers Number One, Employees, and Speed. The CEO detailed Veeva's product evolution, highlighting a strategic shift from a singular focus on Commercial operations to spanning the entire R&D and Commercial continuum. The company initially gained traction starting in 2007 with Veeva CRM, built on the nascent Salesforce.com platform, recognizing its future potential even before features like Apex code were available. While CRM remains a mature product, the primary growth engine has shifted to Veeva Vault, the content management platform and associated applications, which was first sold four years prior to the presentation. The portfolio has expanded further with newer applications like Customer Master and Open Data, a data offering launched two years prior, demonstrating a commitment to becoming a holistic, strategic partner across various buyer types within the life sciences industry. Gassner underscored the importance of maintaining speed and autonomy, even as the company scales, noting that the natural gravitational pull for a large company is toward slowness and bureaucracy. To counteract this, Veeva explicitly accepts a degree of inefficiency, viewing it as a necessary trade-off to maintain agility and empower employees. This approach is tied directly to their strategic goal of achieving a $1 billion run rate by 2020, a five-year target from the time of the presentation. Achieving this ambitious goal requires a significant shift in revenue composition: non-CRM revenue, which accounted for approximately 20% of the run rate, must increase to 50%. This transition necessitates focused execution and daily effort from the entire organization, confirming that the company's future growth is intrinsically linked to the success and adoption of its non-CRM products, particularly Veeva Vault and its data offerings. ### Detailed Key Takeaways * **Strategic Vision and Values:** Veeva's operations are governed by a clear vision—building the industry cloud for life sciences—and three non-negotiable values: putting Customers Number One, prioritizing Employees (as they are a people-based business requiring long-term commitment), and maintaining Speed. * **The Fight Against Organizational Gravity:** As organizations grow, they naturally become slow and bureaucratic. Veeva actively fights this tendency by explicitly accepting inefficiency as a necessary cost to maintain speed and autonomy across its large structure. * **Ambitious Growth Targets:** The company set a challenging goal to grow from a $400 million annual run rate to a $1 billion run rate within five years (by 2020), emphasizing that this requires disciplined execution and daily effort across all departments. * **Revenue Diversification is Key to Future Growth:** To hit the $1 billion target, Veeva must fundamentally shift its revenue mix. Non-CRM products (primarily Vault and data services) needed to increase their contribution from roughly 20% of total revenue to 50%. * **Product Evolution from CRM to Industry Cloud:** Veeva's initial success was built on Veeva CRM, launched in 2007 on the then-nascent Salesforce.com platform. This product is now mature, while Veeva Vault (content management and applications) has become the primary growth engine. * **Holistic Partner Strategy:** To become a truly strategic partner in the life sciences industry, companies must find offerings that appeal to different buyers within the sector. Gassner suggests that optimizing for profit on every single application is counterproductive to building a holistic, strategic relationship. * **Early Platform Adoption:** Veeva demonstrated foresight by building on the Salesforce.com platform in 2007, even before essential development tools like Apex code were available, betting on the platform's future maturity. * **Data Offerings as a Growth Vector:** The introduction of data services like Customer Master and Open Data represents a critical expansion beyond core software applications, providing essential data infrastructure for life sciences commercial operations. * **Financial Transparency and Commitment:** The CEO noted the significance of sharing long-term strategic goals, such as the $1 billion run rate target, with financial analysts and owners, treating it as a public commitment that ensures accountability. ### Tools/Resources Mentioned * **Veeva CRM:** The foundational Customer Relationship Management platform for the pharmaceutical industry. * **Veeva Vault:** The content management platform and application suite, identified as the company's primary growth engine. * **Veeva Customer Master:** An application for managing customer data. * **Veeva Open Data:** A data offering designed to provide reliable, open data to the life sciences industry. * **Salesforce.com:** The platform upon which Veeva CRM was initially built. ### Key Concepts * **Industry Cloud:** The strategic vision of Veeva, representing a specialized, comprehensive suite of cloud applications and data services tailored specifically for the unique needs and regulatory requirements of the pharmaceutical and life sciences sector, spanning R&D through Commercial operations. * **Speed over Efficiency:** A core management philosophy where the company prioritizes rapid execution, autonomy, and agility over strict internal efficiency, recognizing that some redundancy is necessary to avoid the bureaucratic slowdown common in large enterprises. * **Run Rate:** A financial metric used to project annual revenue based on current performance, used here to define the company's financial scale and growth targets (e.g., $400 million run rate moving toward $1 billion).